170.
Y.A. Rodríguez-Lucart, I. Contreras-Saá, C.F. Olguín, M.J. Vera, M. Monroy-Cárdenas, C. Almarza, G. Tapia, R. Pulgar, J. Martínez, J. Caballero, R. Araya-Maturana*, F. A. Urra*
169.
J.L. Valdes-Albuernes, E. Díaz-Pico, J.L. Velázquez–Libera, J. Caballero*.

167.
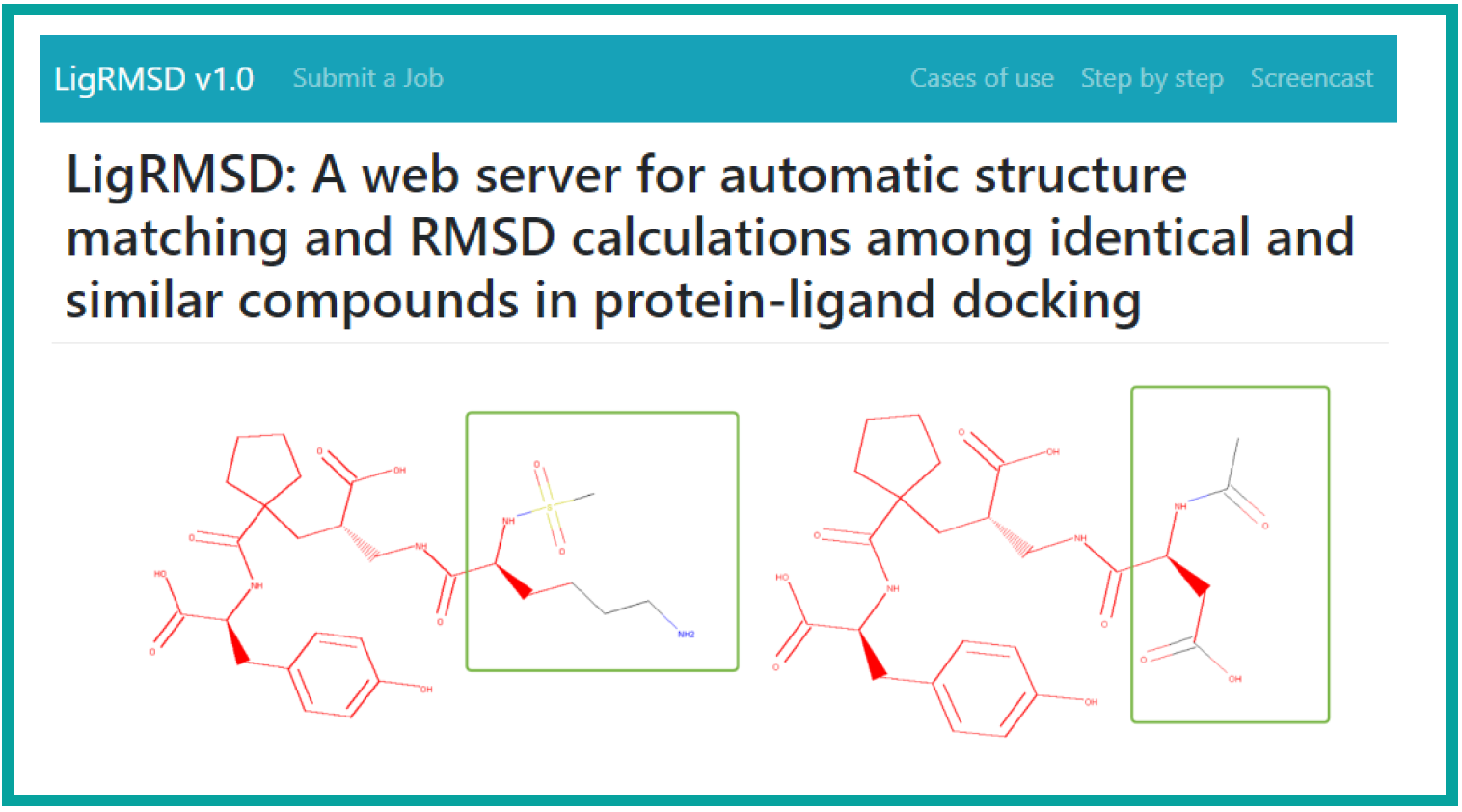
S. Alfaro, F. González-Norambuena, J.L. Velázquez–Libera, F. Adasme-Carreño*, J. Caballero*.
165.
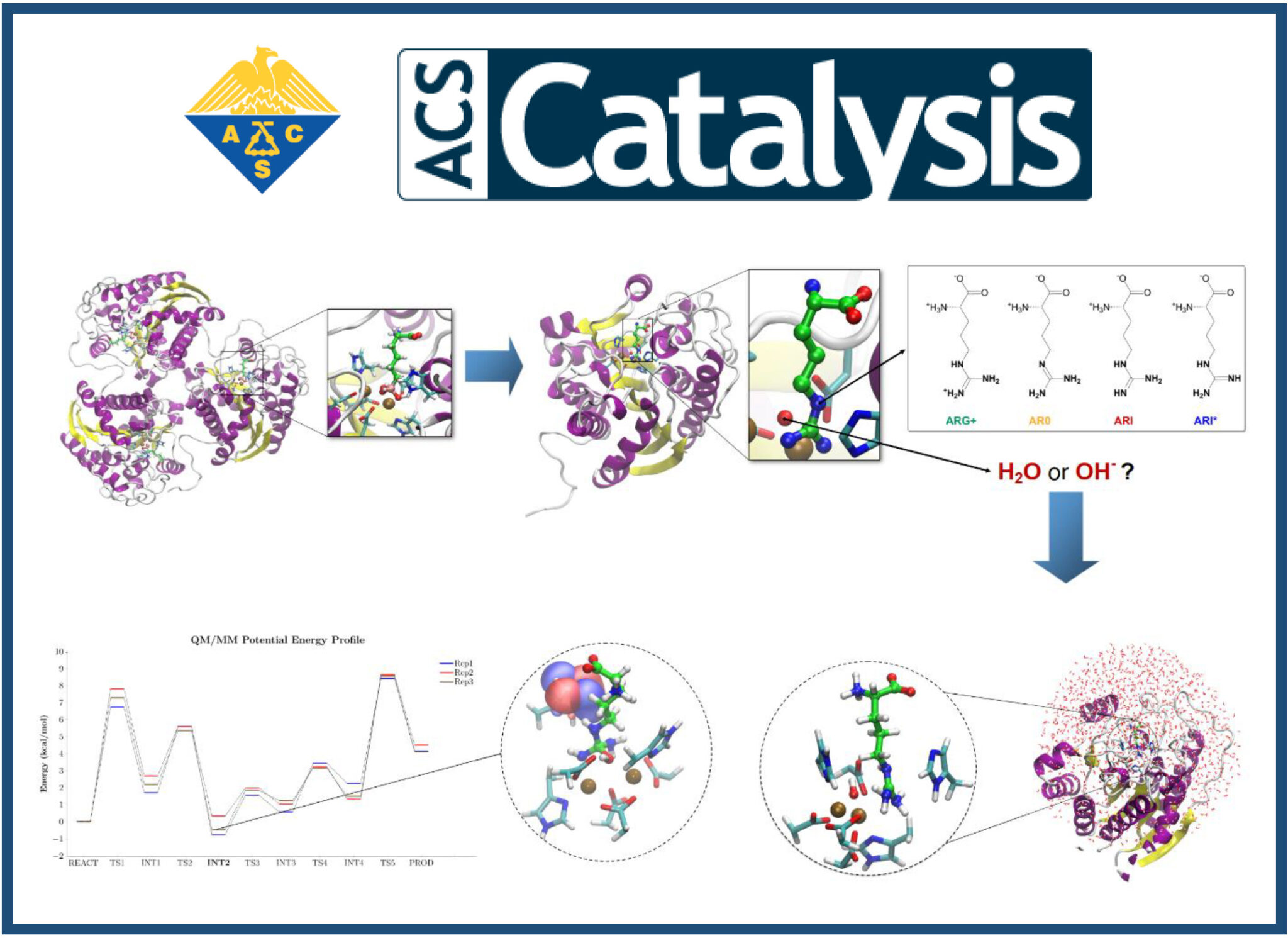
J.L. Velázquez–Libera*, R. Recabarren, E. Vöhringer-Martinez, Y. Salgueiro, J.J. Ruiz-Pernía, J. Caballero, I. Tuñón.
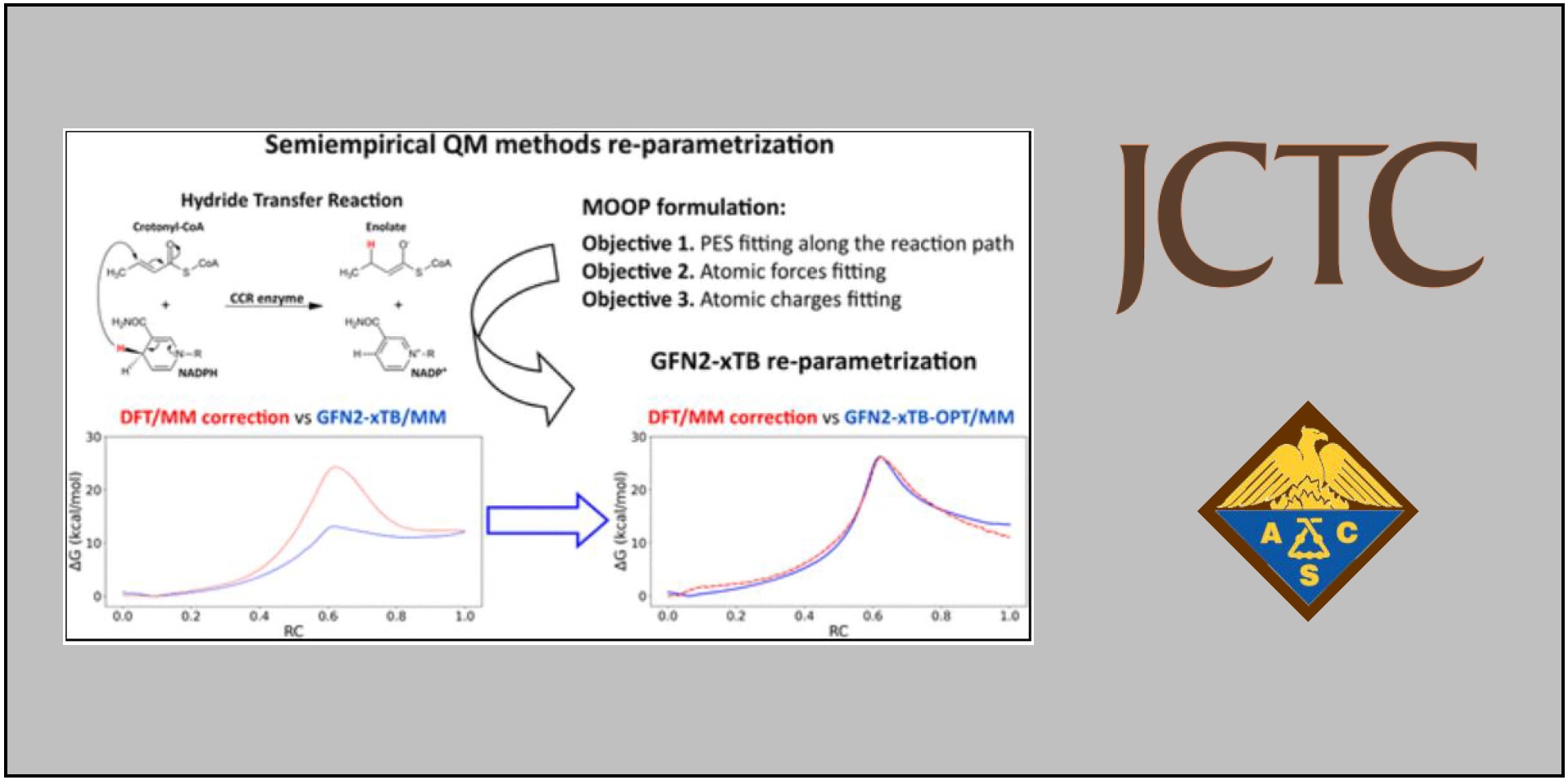
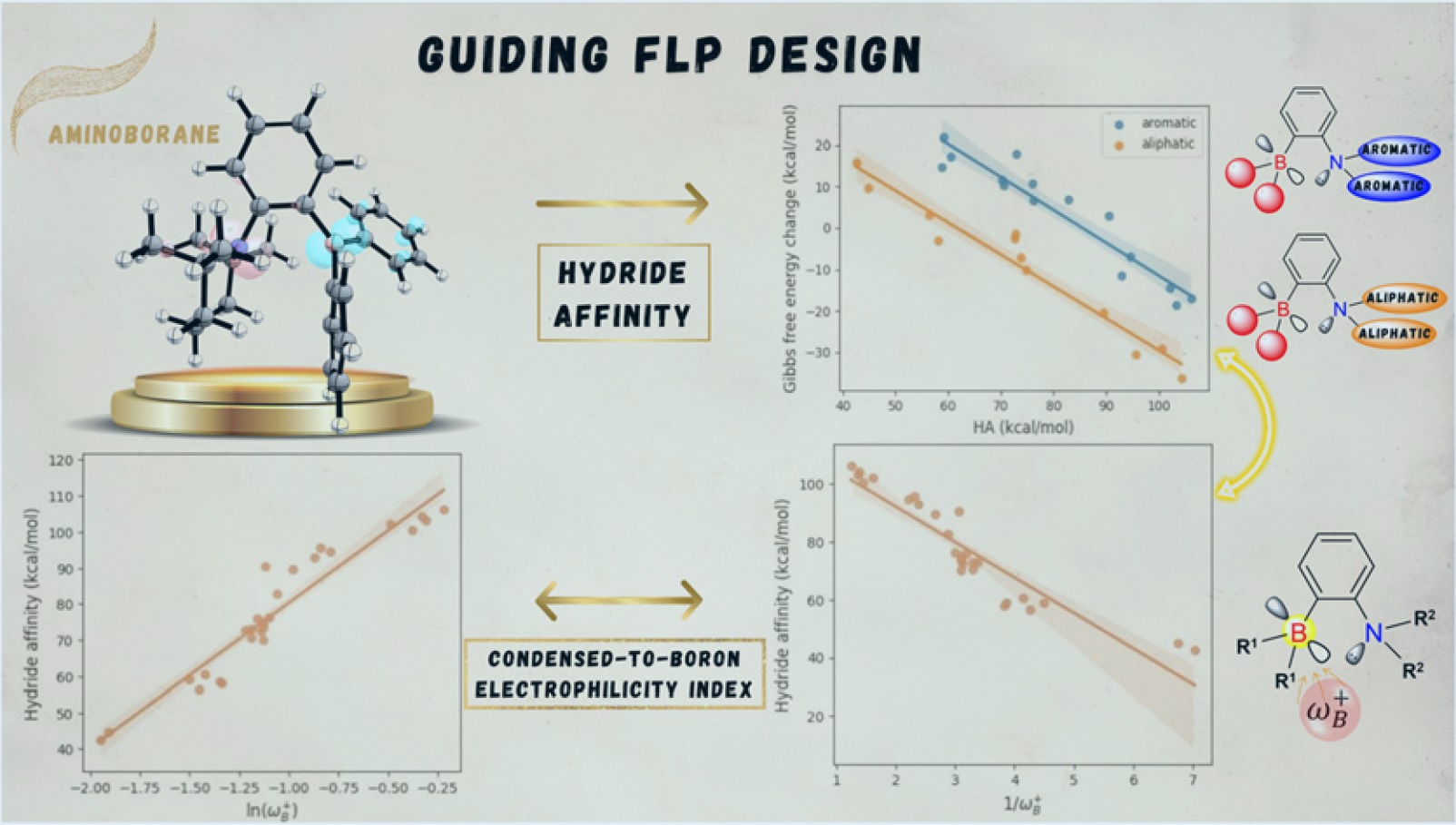
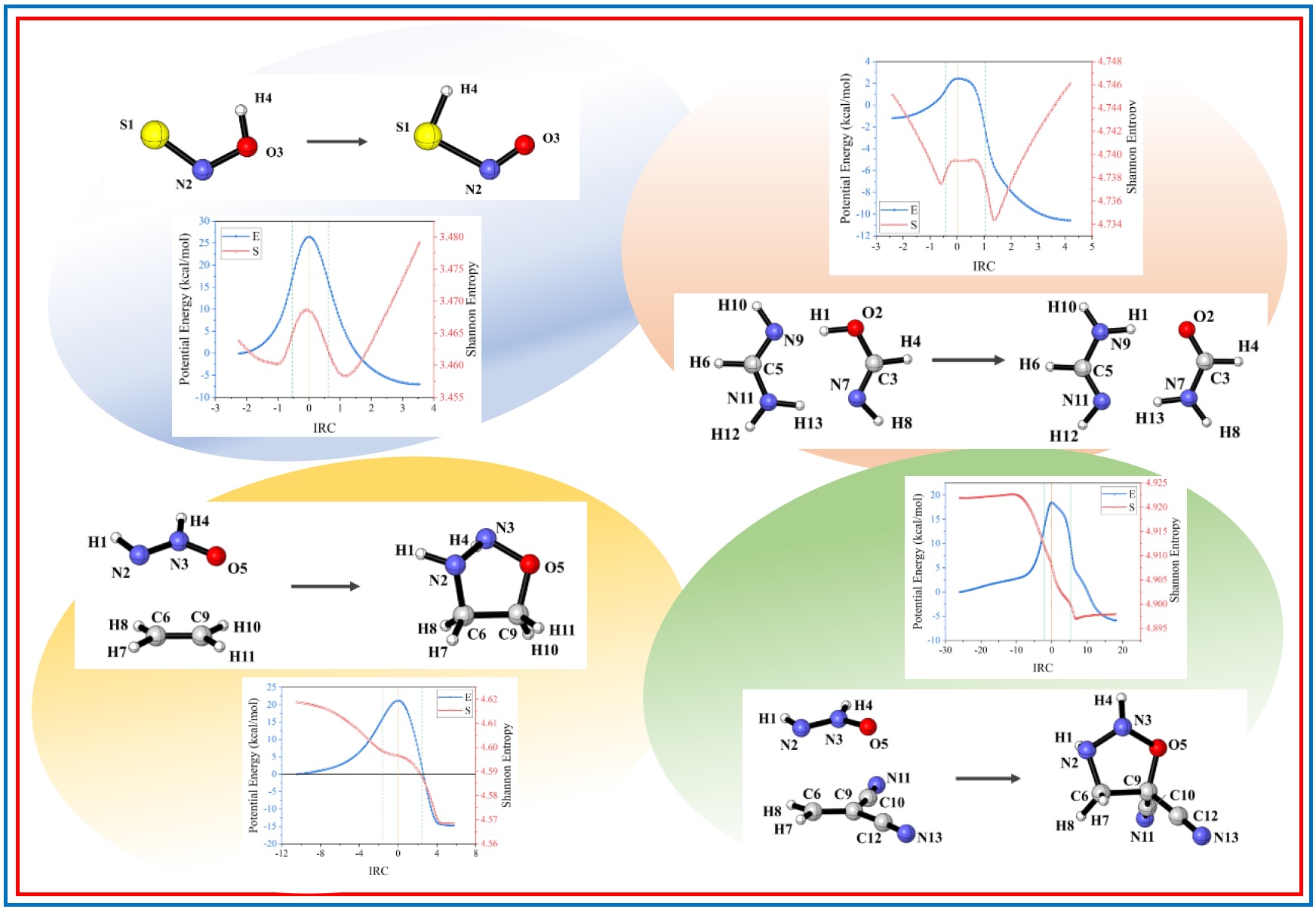
162.
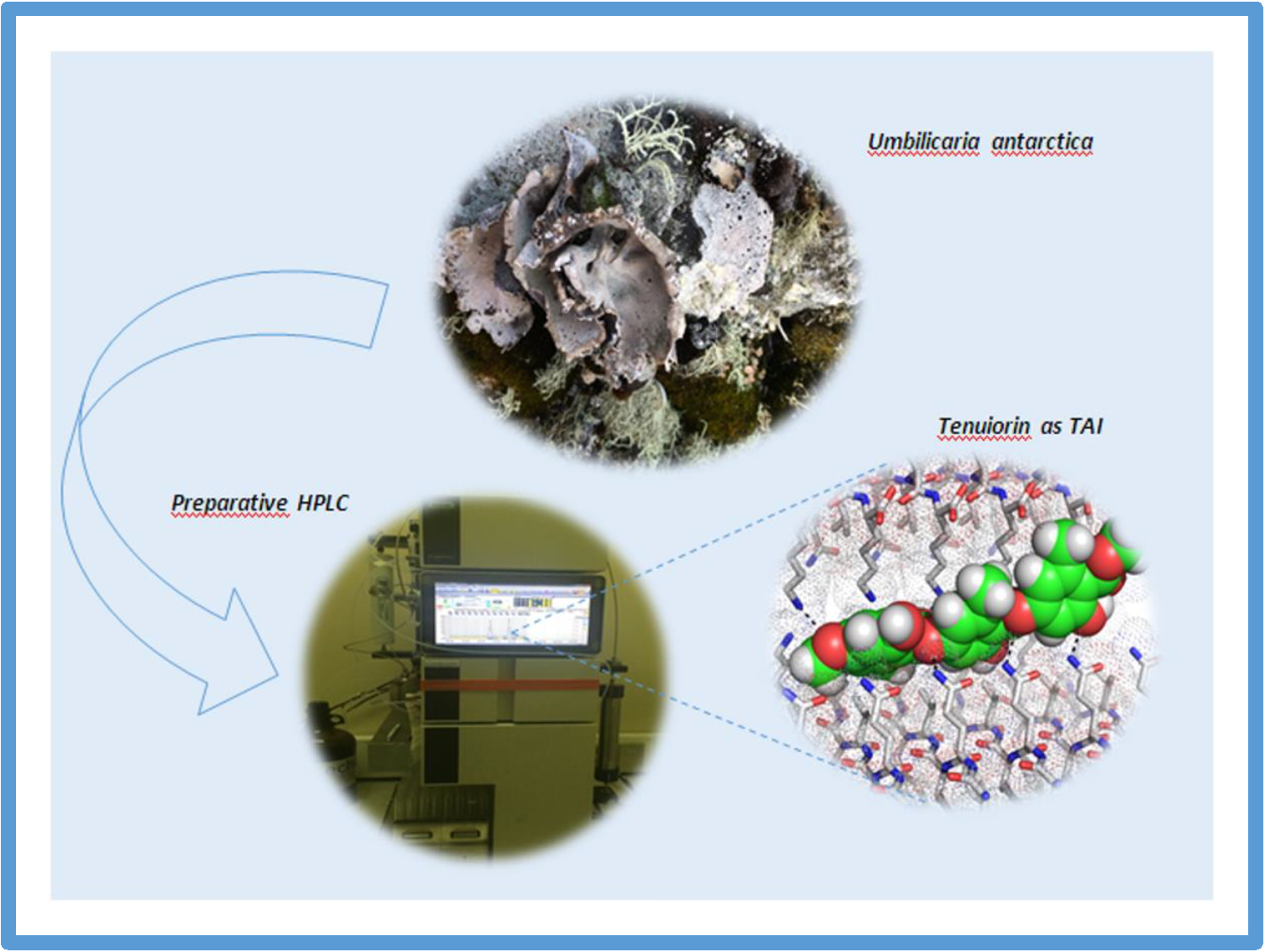
N. Flores, L. Rivillas-Acevedo, J. Caballero, F. Melo, L. Caballero, C. Areche, D. Fuentealba, F. Aguilar, A. Cornejo*.
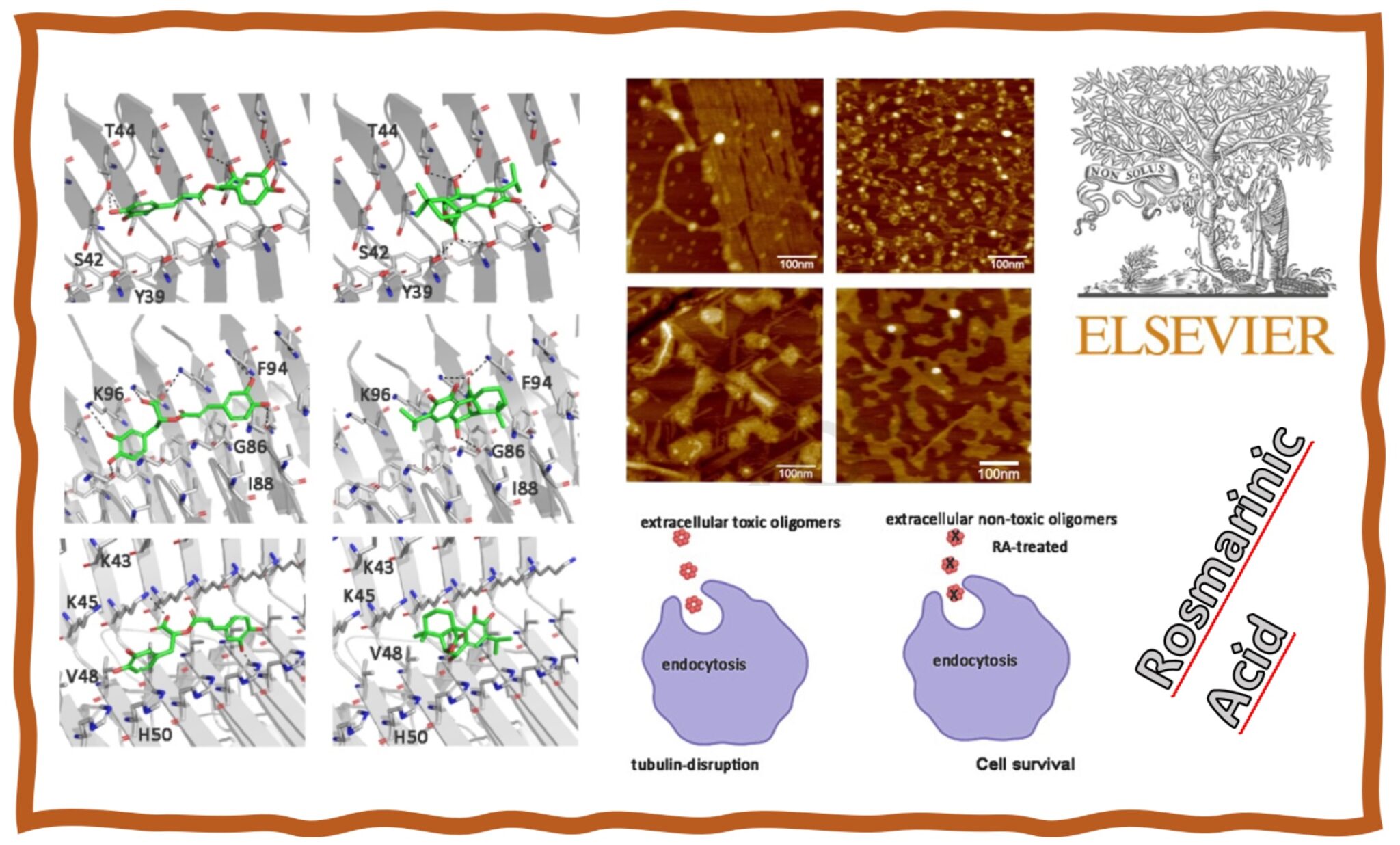
161.
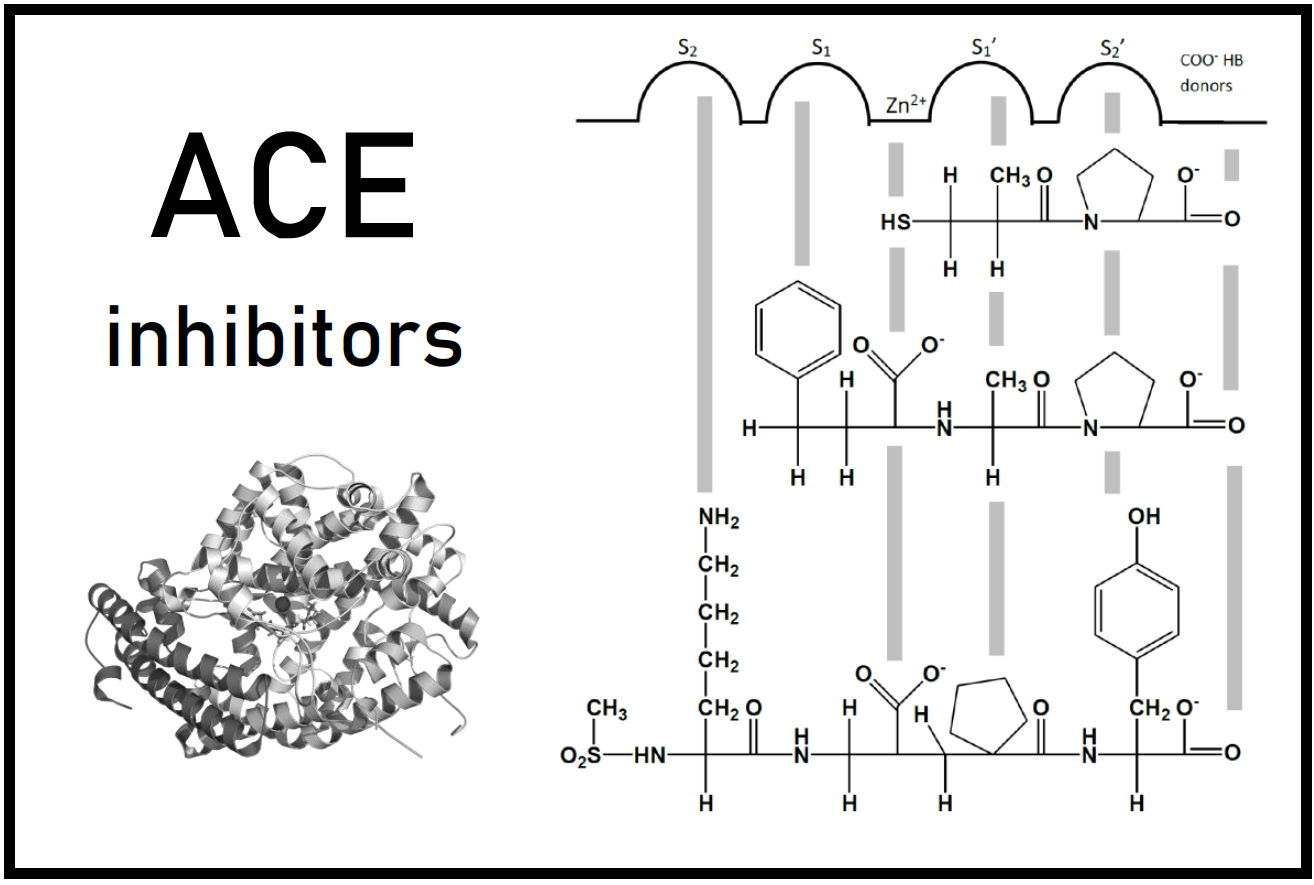
J.L. Velázquez–Libera*, J. Caballero*, J. Alzate-Morales, J.J. Ruiz-Pernía, I. Tuñón*.
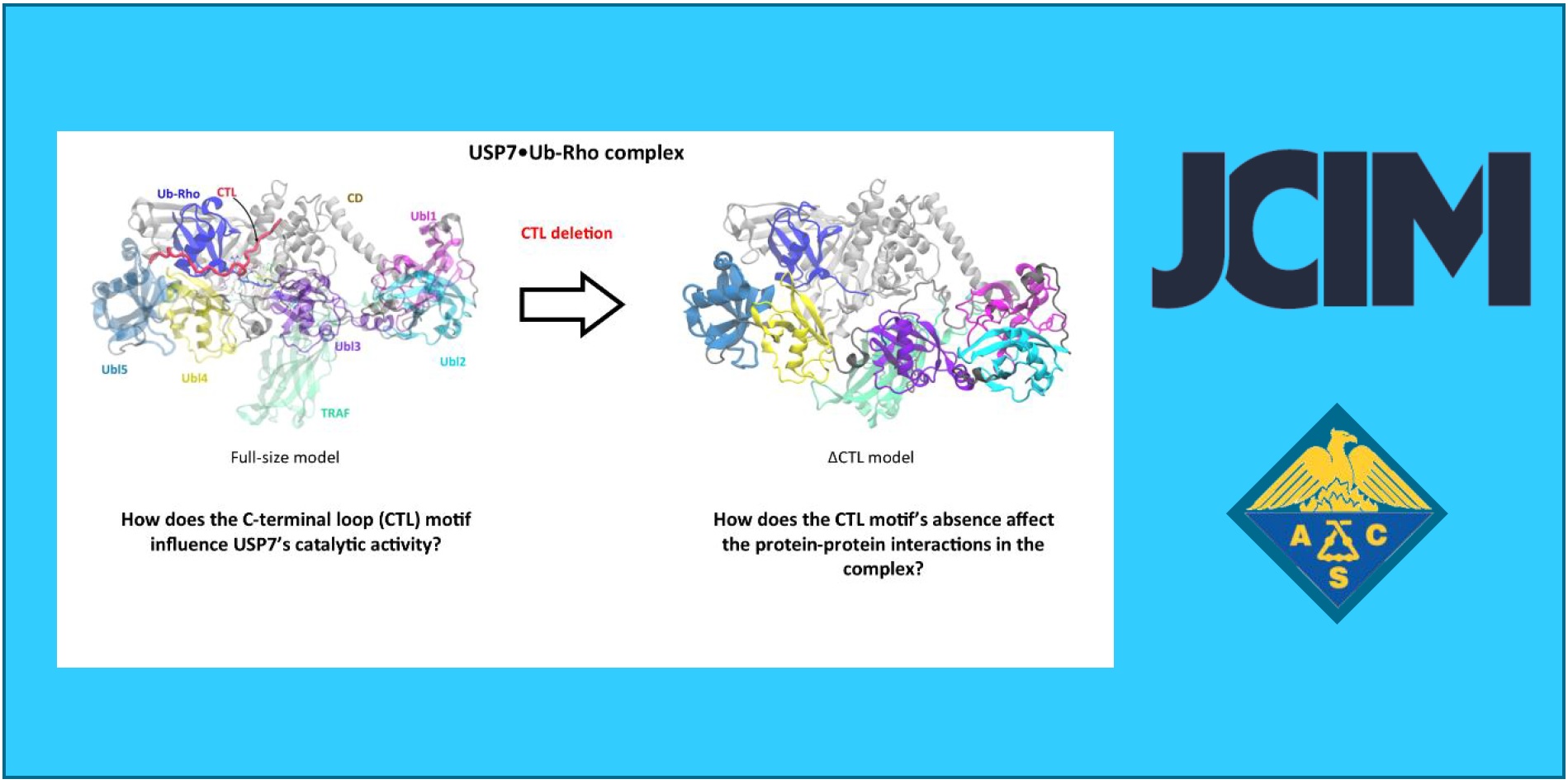
159.
D. Méndez, F. Tellería, M. Monroy-Cárdenas, H. Montecino-Garrido, S. Mansilla, L. Castro, A. Trostchansky, F. Muñoz-Córdova, V. Zickermann, J. Schiller, S. Alfaro, J. Caballero, R. Araya-Maturana*, E. Fuentes*
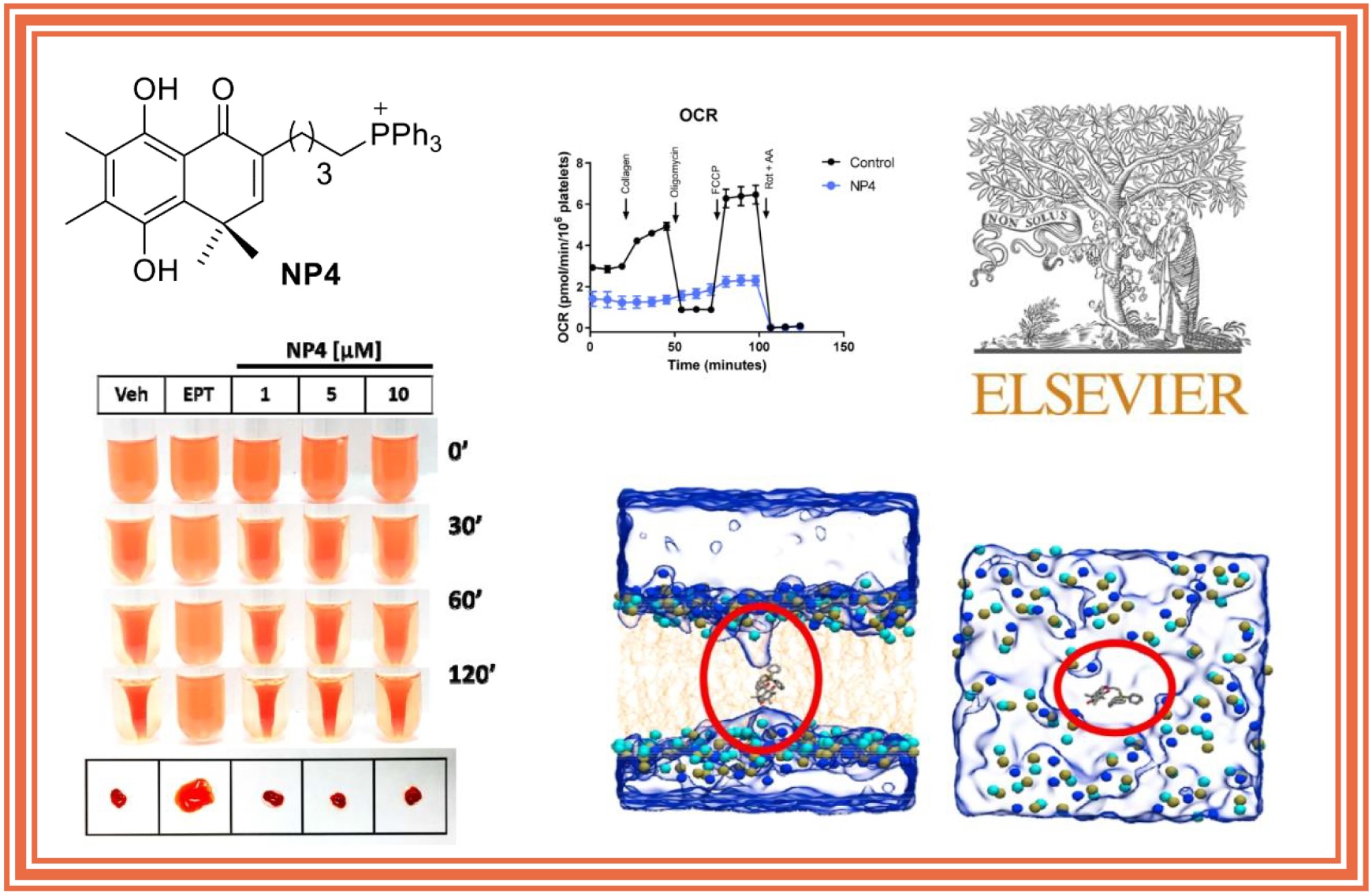
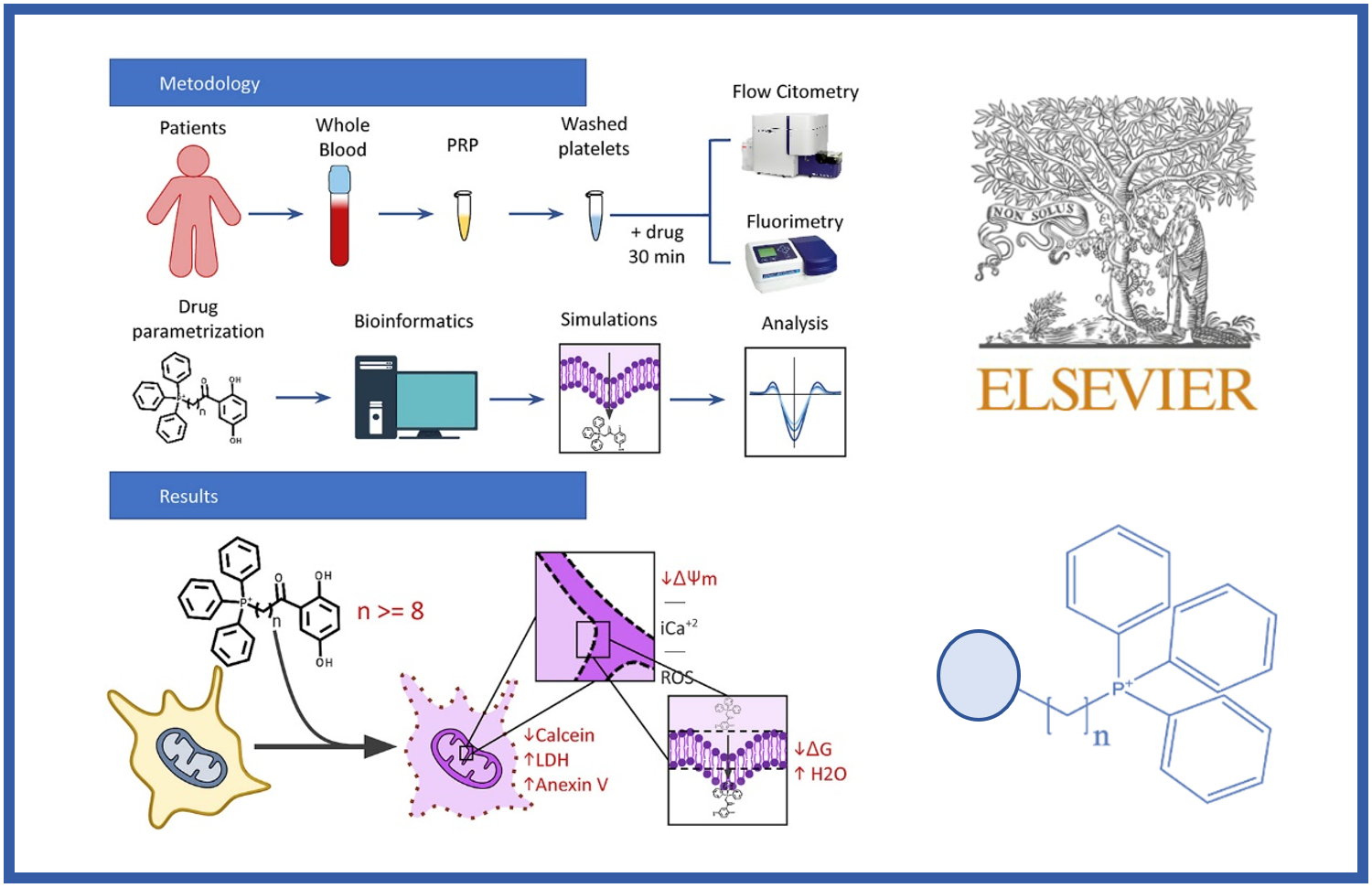
157.
H. Montecino-Garrido, M. Sepúlveda, D. Méndez, M. Monroy-Cárdenas, S. Alfaro, M. González-Avendaño, J. Caballero, F.A. Urra, R. Araya-Maturana*, E. Fuentes*.
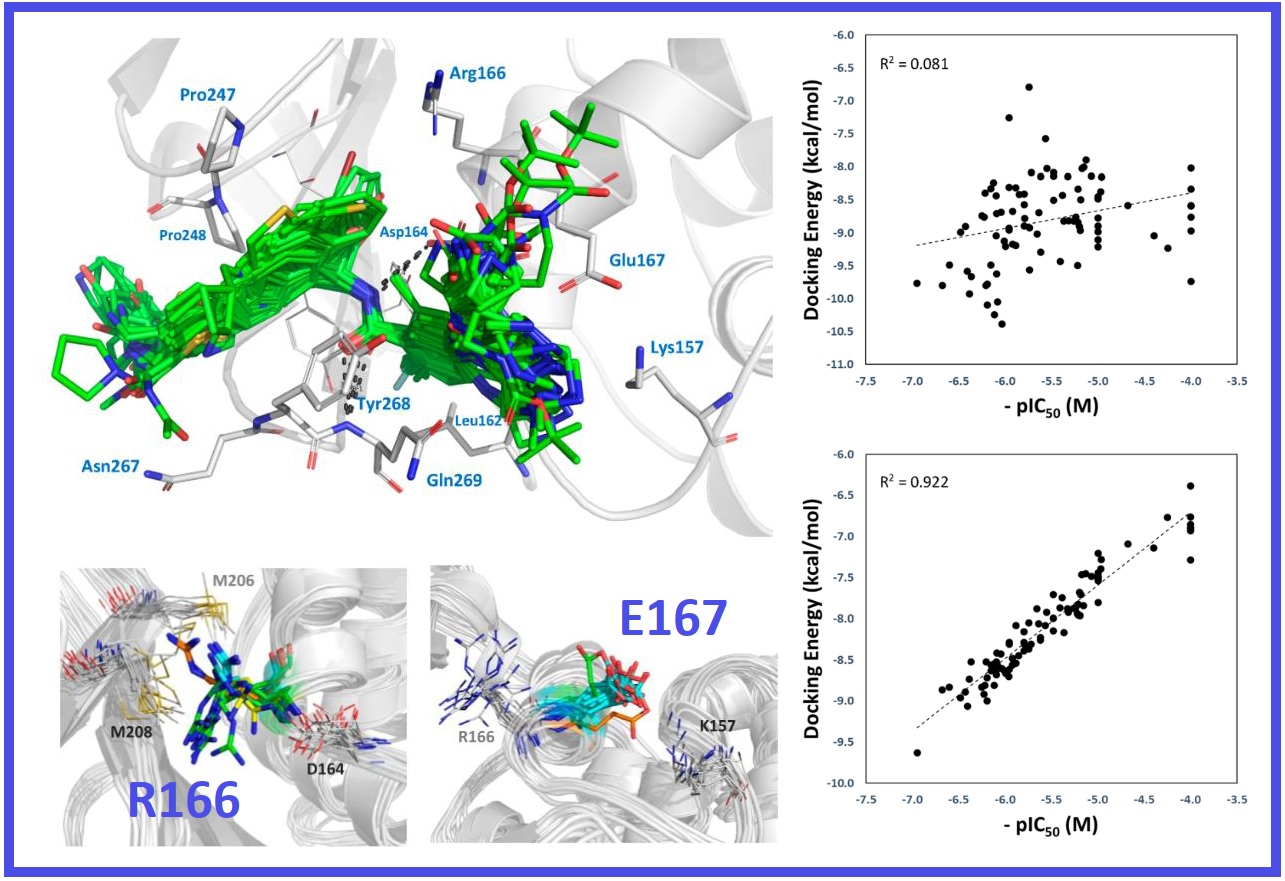
156.
R. González, J. Murillo-López, W. Rabanal-León, L. Prent-Peñaloza, O. Concepción, P. Olivares, Y. Duarte, A.F. de la Torre, M. Gutiérrez*, J. Caballero*.
155.
J.L. Velázquez–Libera*, J. Caballero*, J. Alzate-Morales, J.J. Ruiz-Pernía, I. Tuñón*.
153.
S. Monroy-Moya, J. Caballero, F. González-Norambuena, M. Simirgiotis, E. Sánchez, C. Areche, D. Fuentealba, A. Cornejo*.
152.
R. González-Alemán*, D. Platero-Rochart, A. Rodríguez-Serradet, E.W. Hernández-Rodríguez, J. Caballero, F. Leclerc*, L. Montero-Cabrera.
151.
I. Monte*, J. Caballero, A.M. Zamarreño, G. Fernández-Barbero, J.M. García-Mina, R. Solano*.
149.
D. Platero-Rochart*, R. González-Alemán*, E.W. Hernández-Rodríguez, F. Leclerc, J. Caballero, L. Montero-Cabrera.
148.
R. González-Alemán*, D. Platero-Rochart, D. Hernández-Castillo, E.W. Hernández-Rodríguez, J. Caballero, F. Leclerc, L. Montero-Cabrera.
147.
G. Rossino, M. Rui, P. Linciano, D. Rossi, M. Boiocchi, M. Peviani, E. Poggio, D. Curti, D. Schepmann, B. Wünsch, M. González-Avendaño, A. Vergara-Jaque, J. Caballero*, S. Collina*.
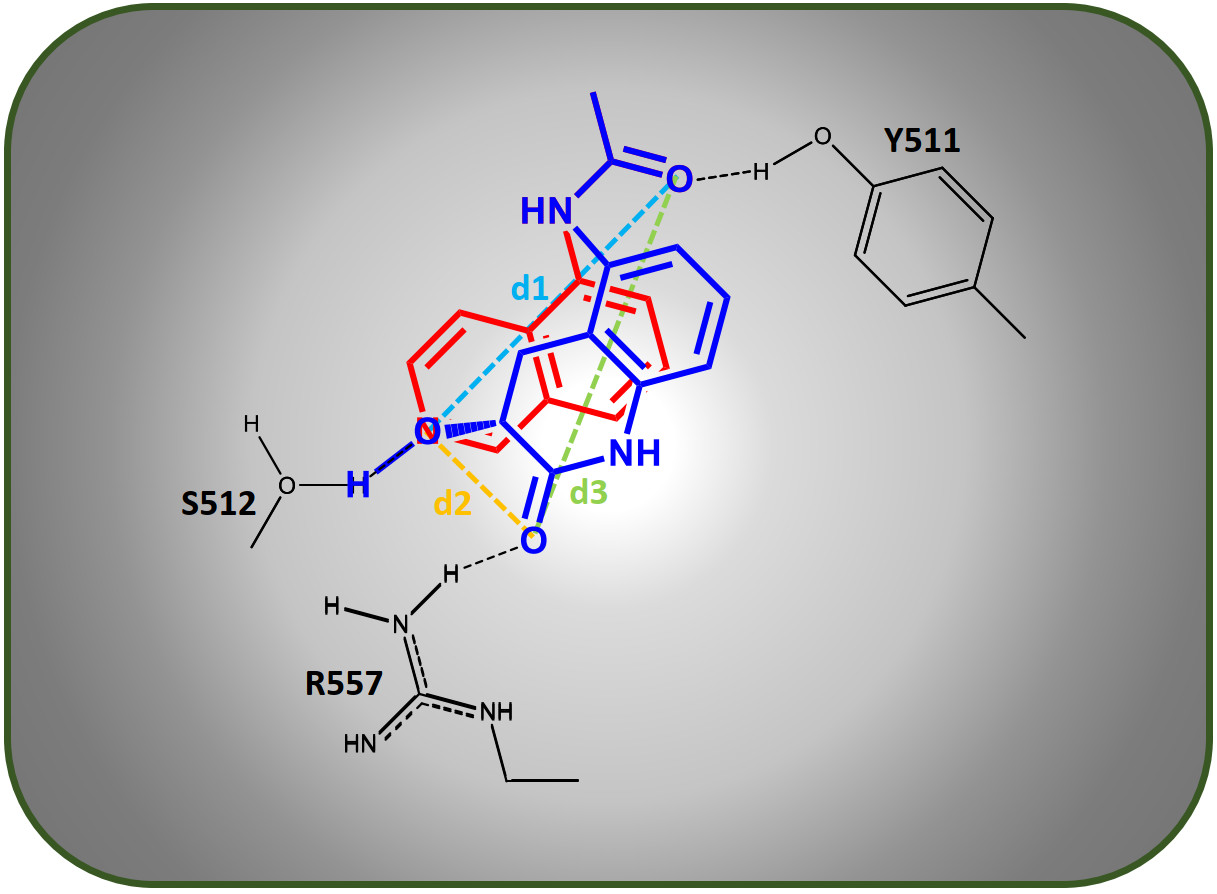
143.
A. Cornejo*, J. Caballero, M. Simirgiotis, V. Torres, L. Sánchez, N. Díaz, M. Guimaraes, M. Hernández, C. Areche, S. Alfaro, L. Caballero, F. Melo.
142.
F. Klein, D. Cáceres, M. Carrasco, J.C. Tapia, J. Caballero, J. Alzate-Morales, S. Pantano*
141.
E.W. Hernández-Rodríguez*, A.M. Escorcia, M.W. van der Kamp, A.L. Montero-Alejo, J. Caballero.
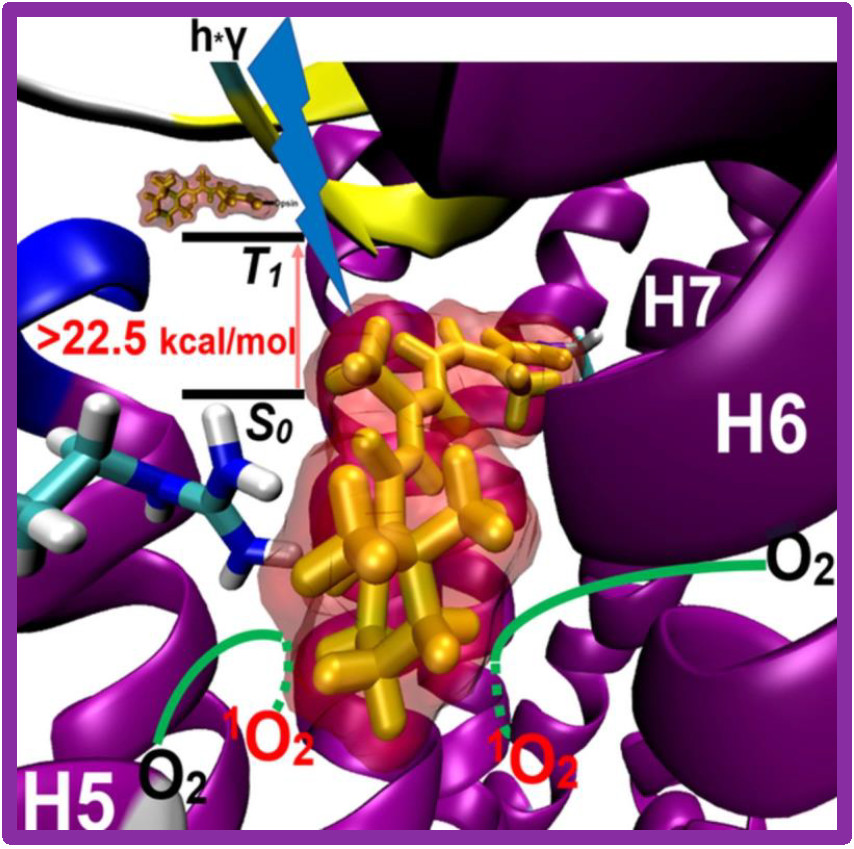
140.
J.L. Velázquez–Libera, J. Caballero*, I. Tuñón*, E.W. Hernández-Rodríguez*, J.J. Ruiz-Pernía.
138.
J.L. Velázquez–Libera, F. Durán-Verdugo, A. Valdés-Jiménez, G. Núñez-Vivanco*, J. Caballero*
137.
F. Salgado, M. Silva, J. Caballero, R. Vargas, A. Cornejo, C. Areche*
136.
G. Rossino, I. Orellana, J. Caballero, D. Schepmann, B. Wünsch, M. Rui, D. Rossi, M. González-Avendaño, S. Collina*, A. Vergara-Jaque*
135.
R. González-Alemán*, D. Hernández-Castillo, J. Caballero*, L. Montero-Cabrera.
134.
R. González-Alemán*, D. Hernández-Castillo, A. Rodríguez-Serradet, J. Caballero, E.W. Hernández-Rodríguez, L. Montero-Cabrera.
132.
P. Santos, F. Lopez-Vallejo*, D. Ramírez, J. Caballero, Dulce M. Espinosa, R. Hernández-Pando, C.Y. Soto.
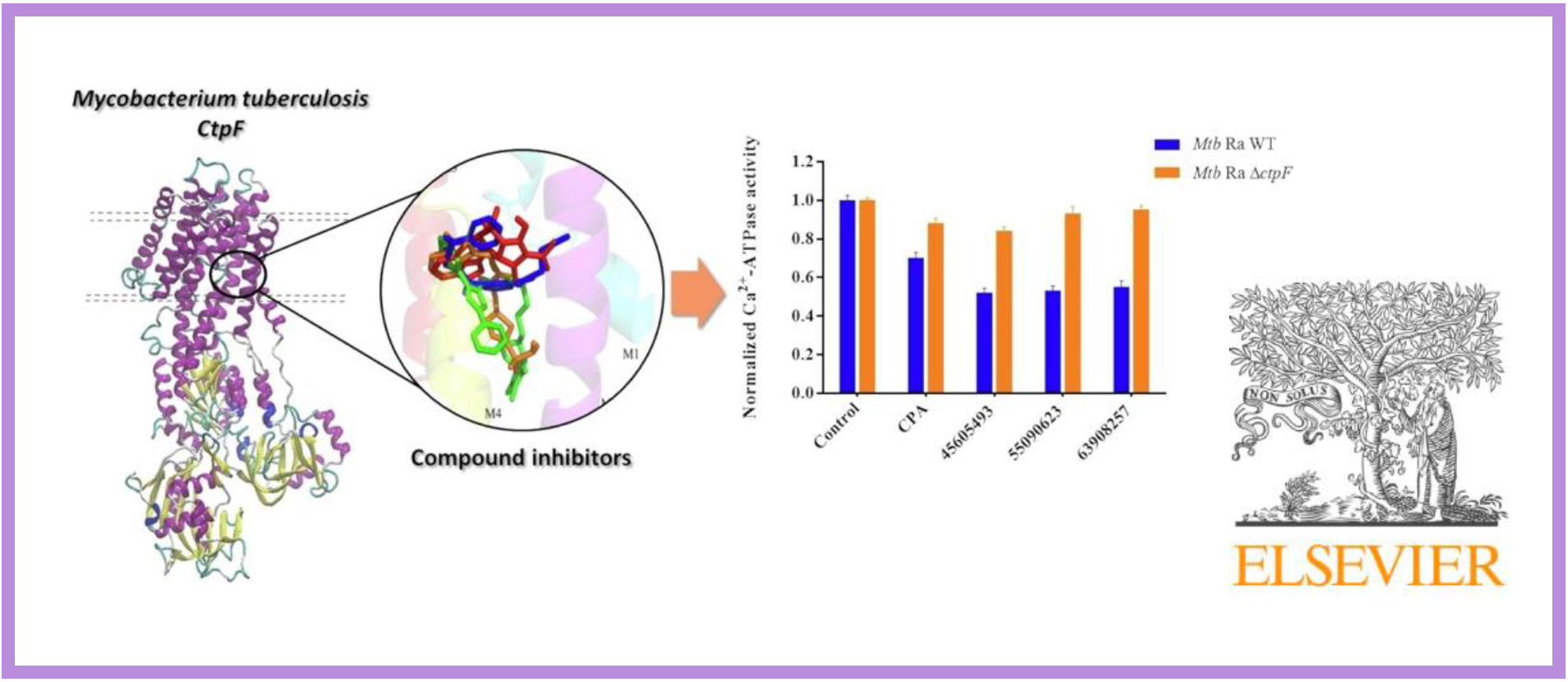
131.
J.L. Velázquez–Libera, J.A. Murillo–López, A.F. de la Torre*, J. Caballero*
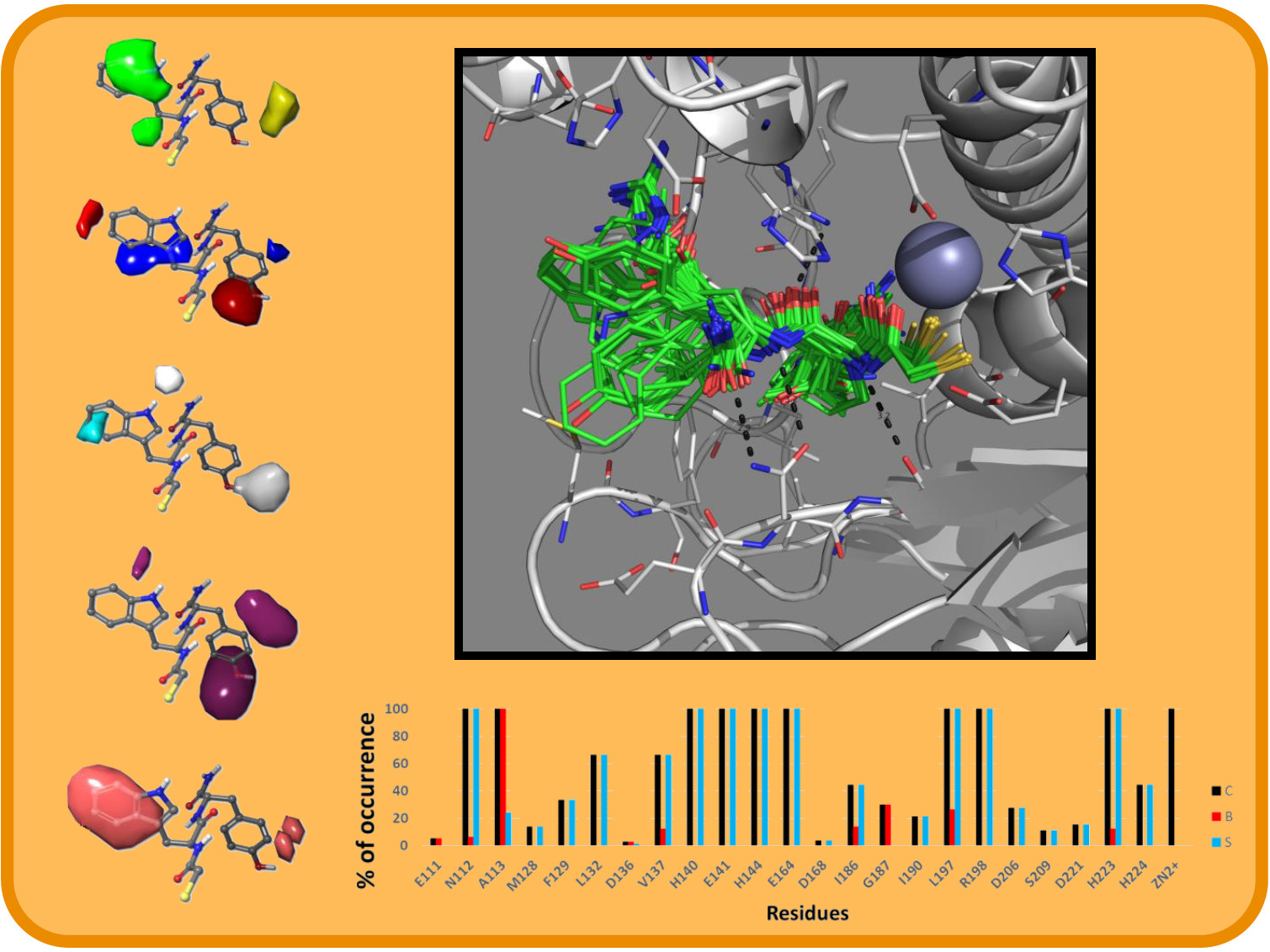
130.
G.H. Jimenez-Aleman*, M. Almeida-Trapp, G. Fernández-Barbero, S. Gimenez-Ibanez, M. Reichelt, J. Vadassery, A. Mithöfer, J. Caballero, W. Boland, R. Solano
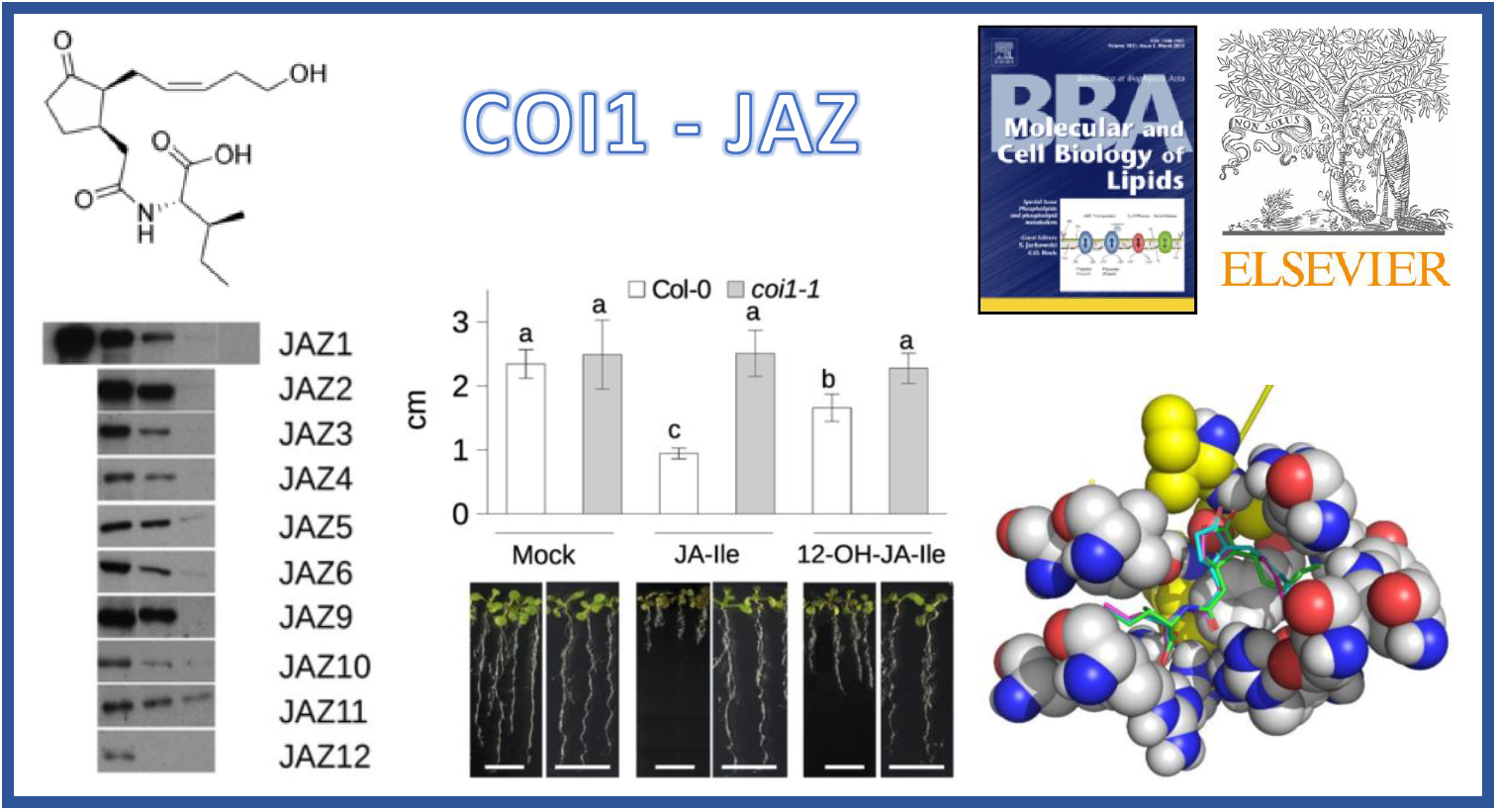
129.
R. Recabarren, E. Osorio, J. Caballero, I. Tuñón*, J. Alzate-Morales*
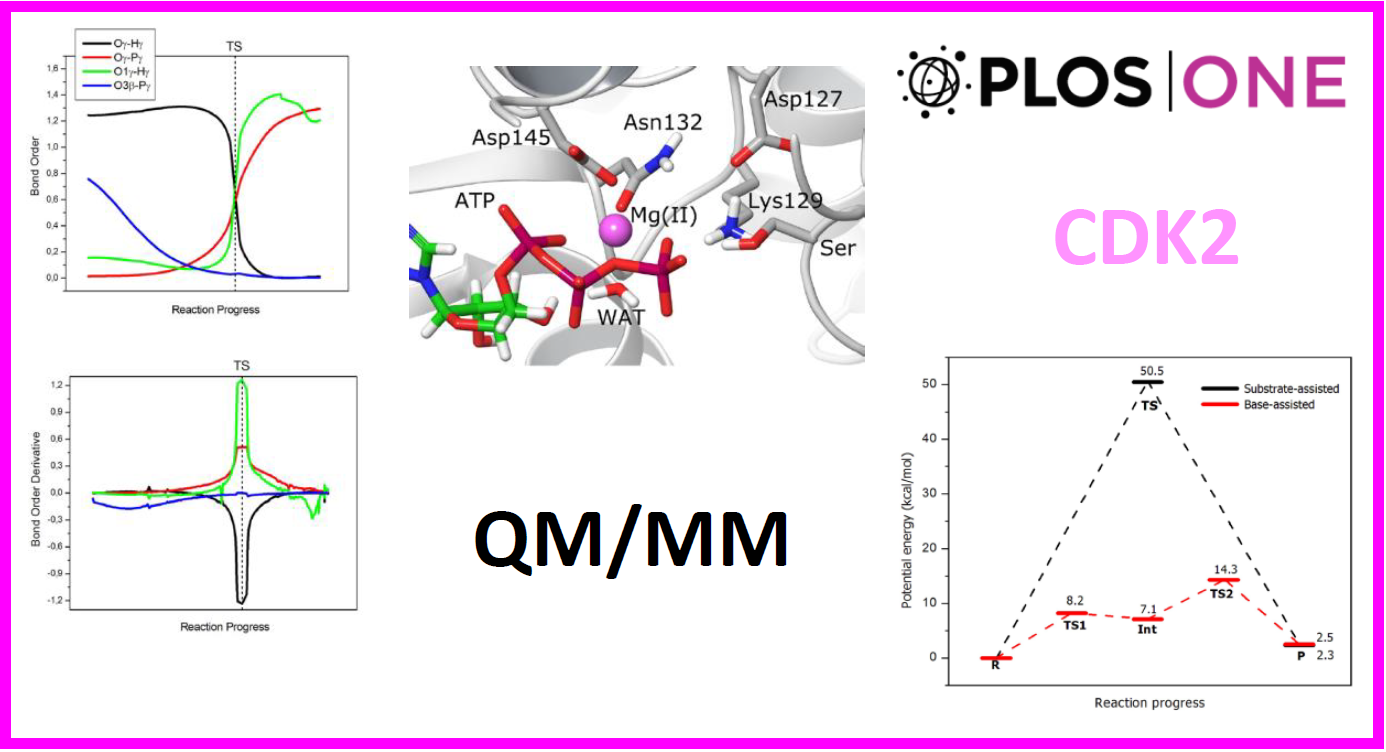
128.
D. Ramírez, G. Concha, B. Arévalo, L. Prent-Peñaloza, L. Zúñiga, A.K. Kiper, S. Rinné, M. Reyes-Parada, N. Decher, W. González*, J. Caballero*
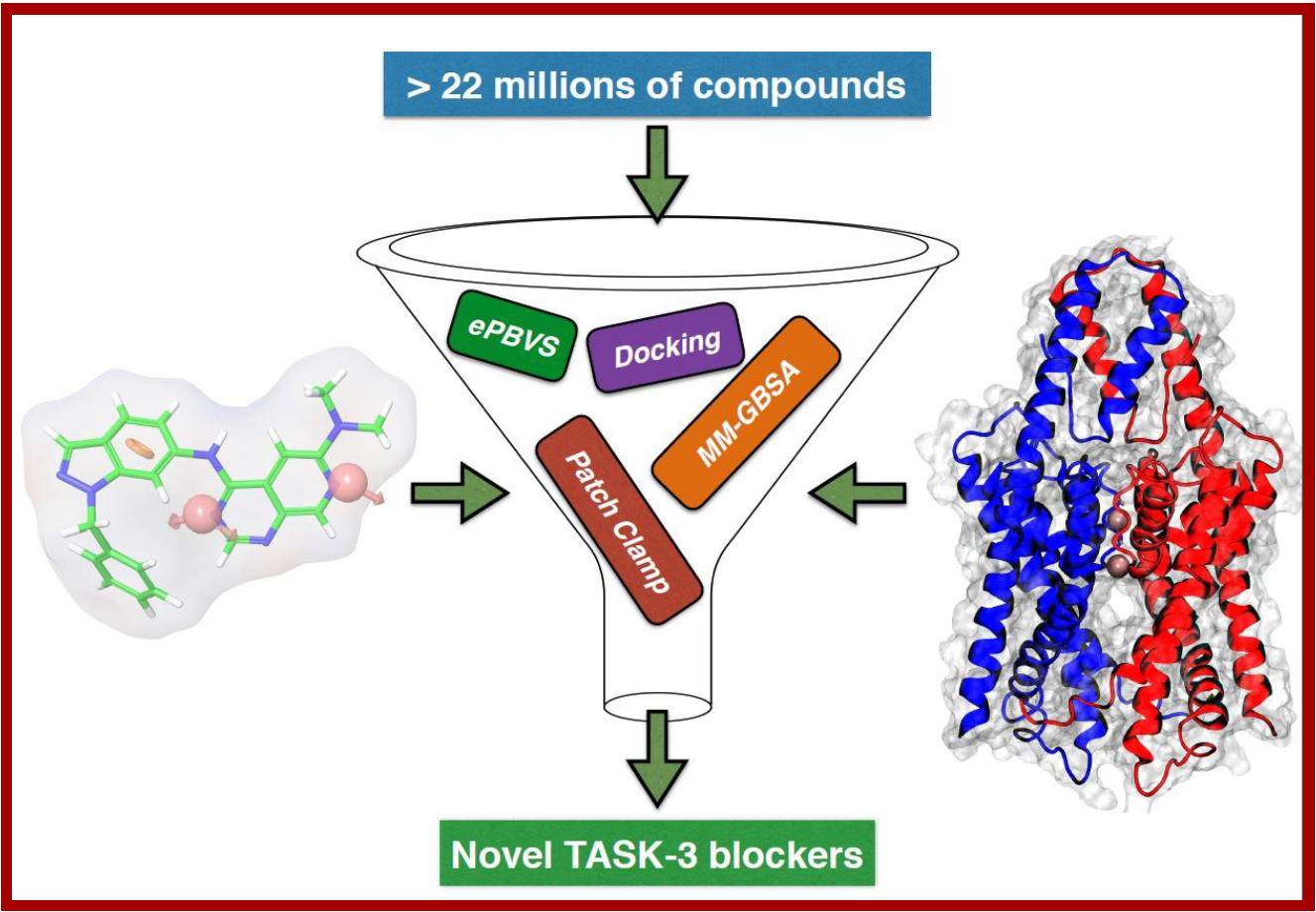
127.
A.F. de la Torre*, A. Ali, O. Concepcion, A.L. Montero-Alejo, F.M. Muñiz, C.A. Jiménez, J. Belmar, J.L. Velázquez–Libera, E.W. Hernández–Rodríguez*, J. Caballero.
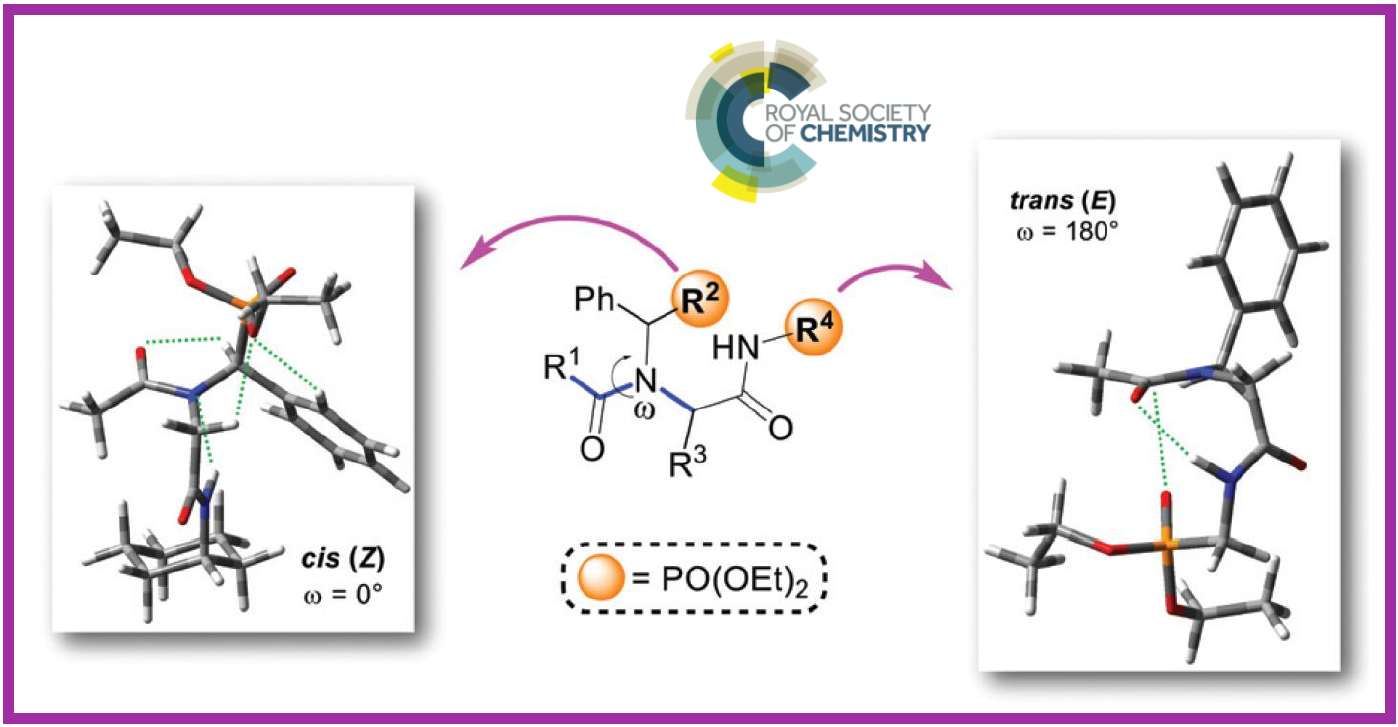
126.
J.L. Velázquez-Libera, G. Rossino, C. Navarro-Retamal, S. Collina, J. Caballero*
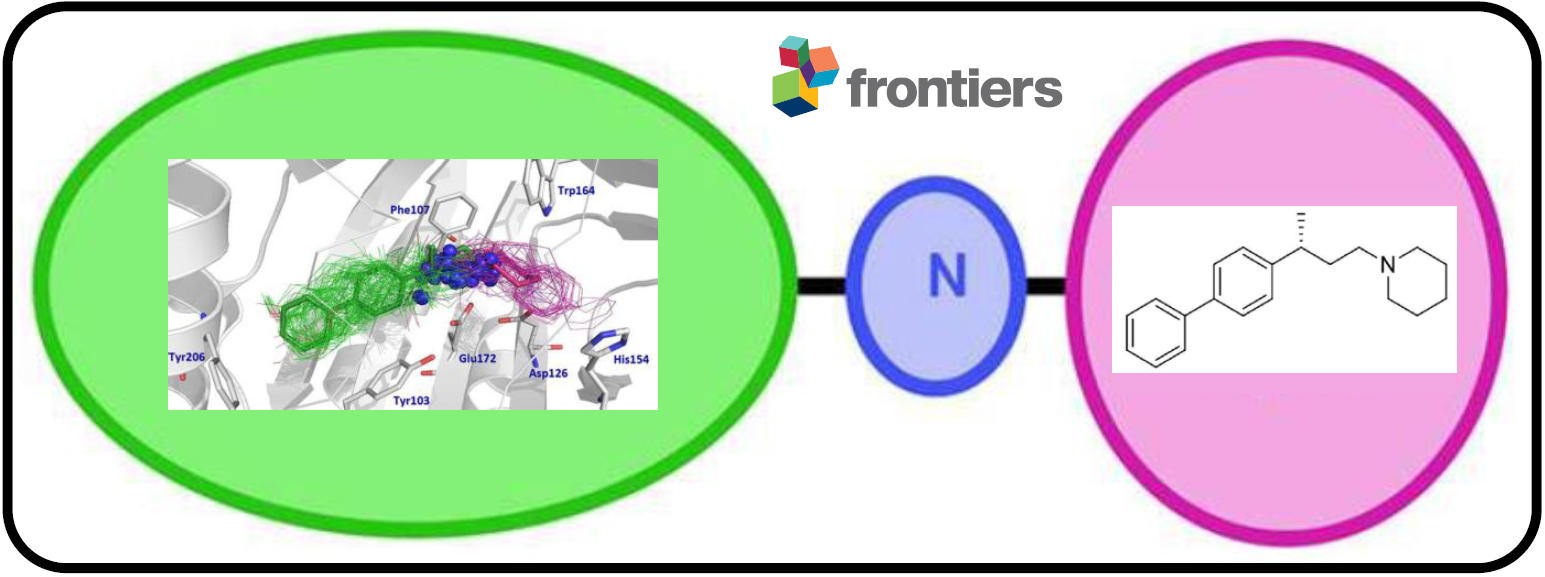
125.
M.N. Tahir, M. Ashfaq, A.F. de la Torre, J. Caballero, E.W. Hernández-Rodríguez*, A. Ali*
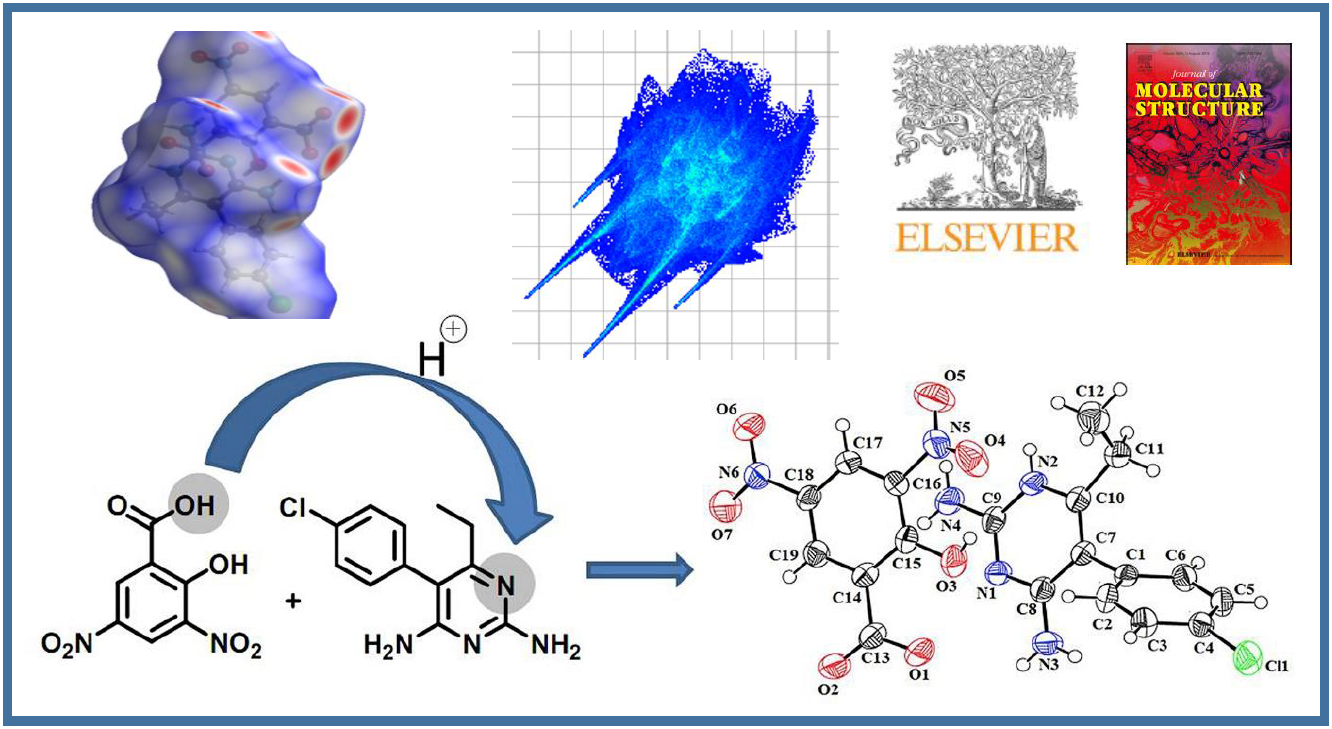
124.
B. Benso, D. Bustos, M.O. Zarraga, W. Gonzalez, J. Caballero, S.E. Brauchi*
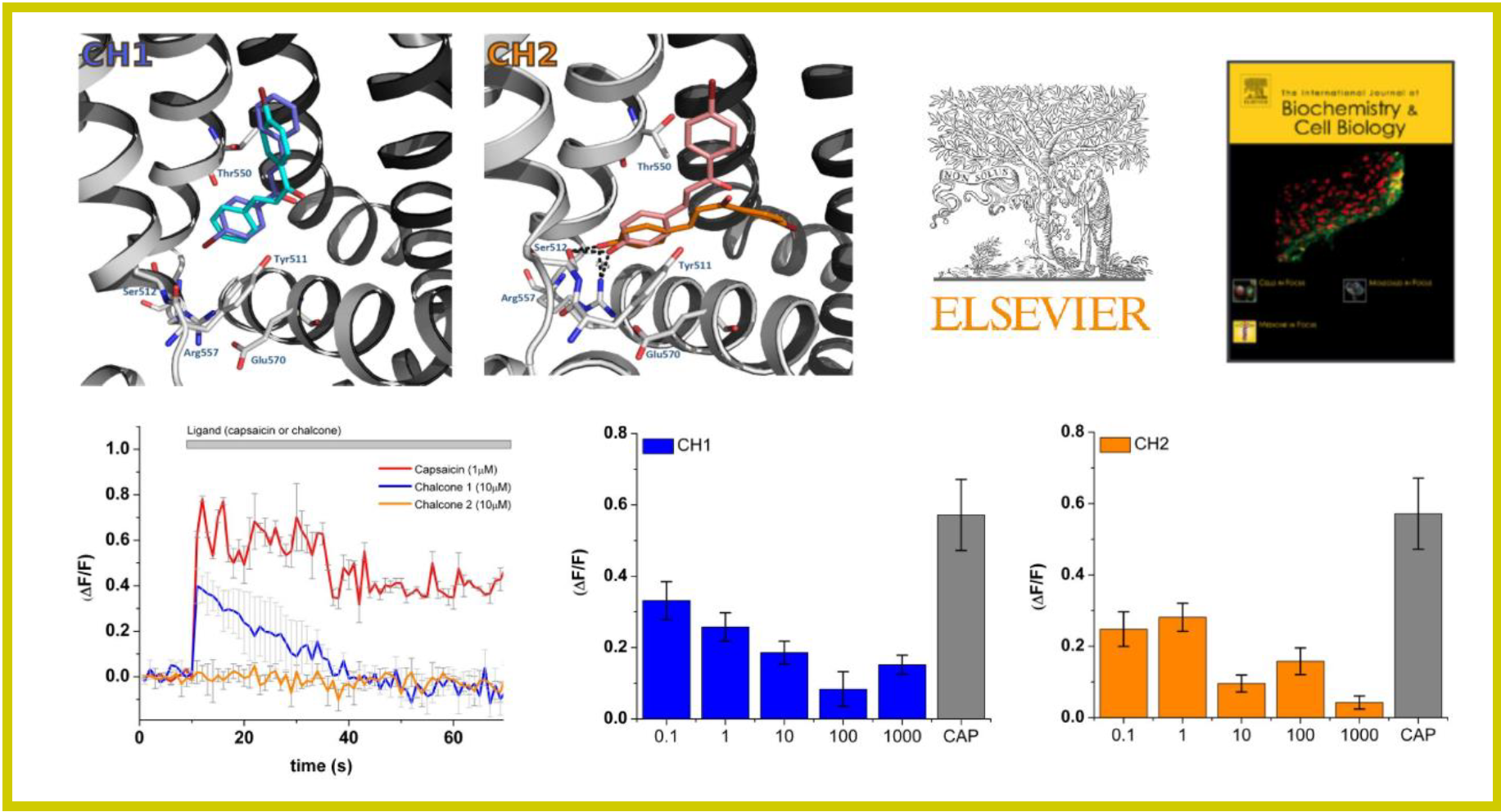
123.
J. Murillo–López, K. Zinovjev, H. Pereira, A. Caniuguir, R. Garratt, J. Babul, R. Recabarren, J. Alzate-Morales, J. Caballero*, I. Tuñón*, R. Cabrera*
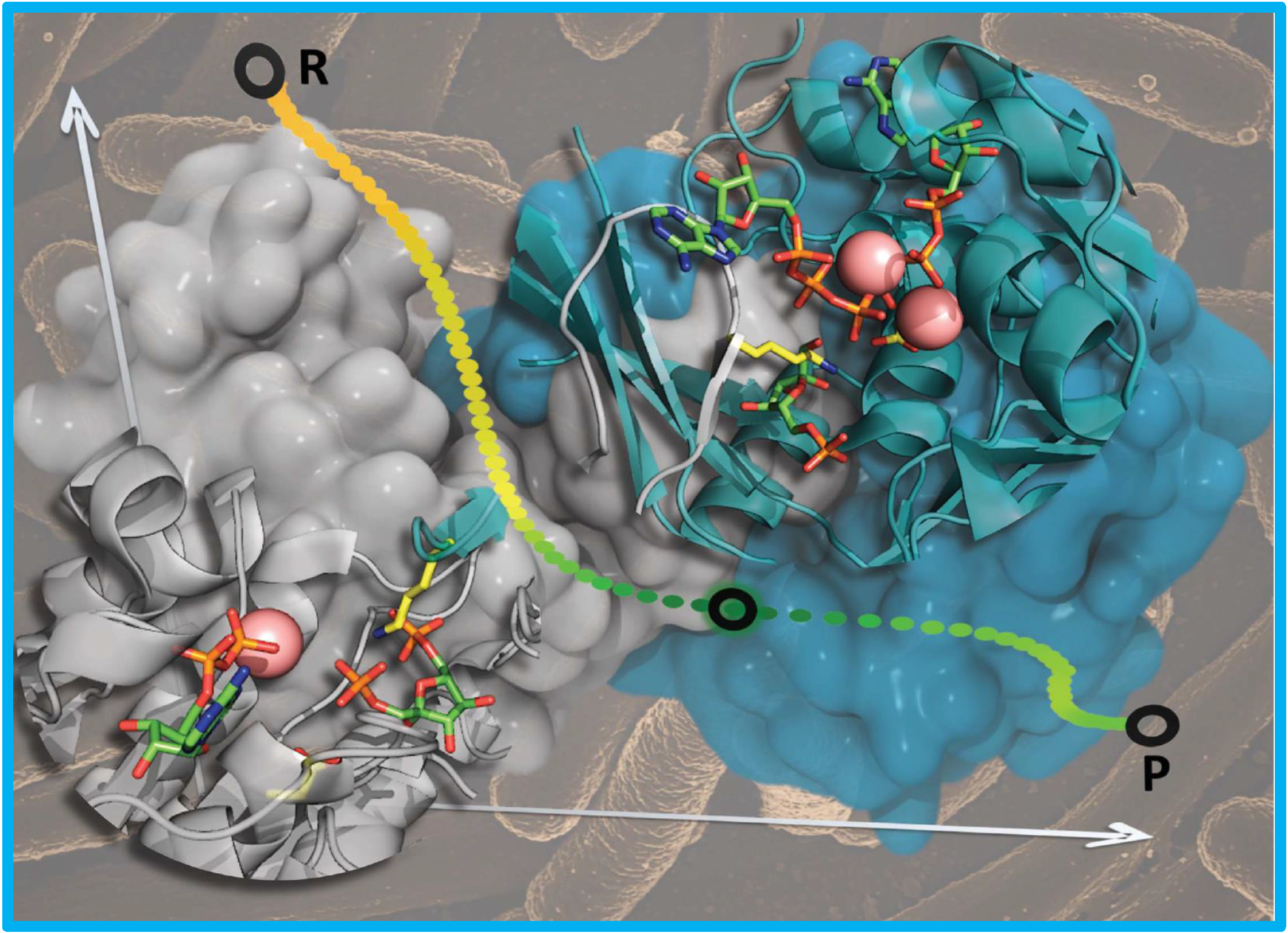
122.
L. Prent-Peñaloza, A.F. de la Torre, J.L. Velázquez-Libera, M. Gutiérrez, J. Caballero*
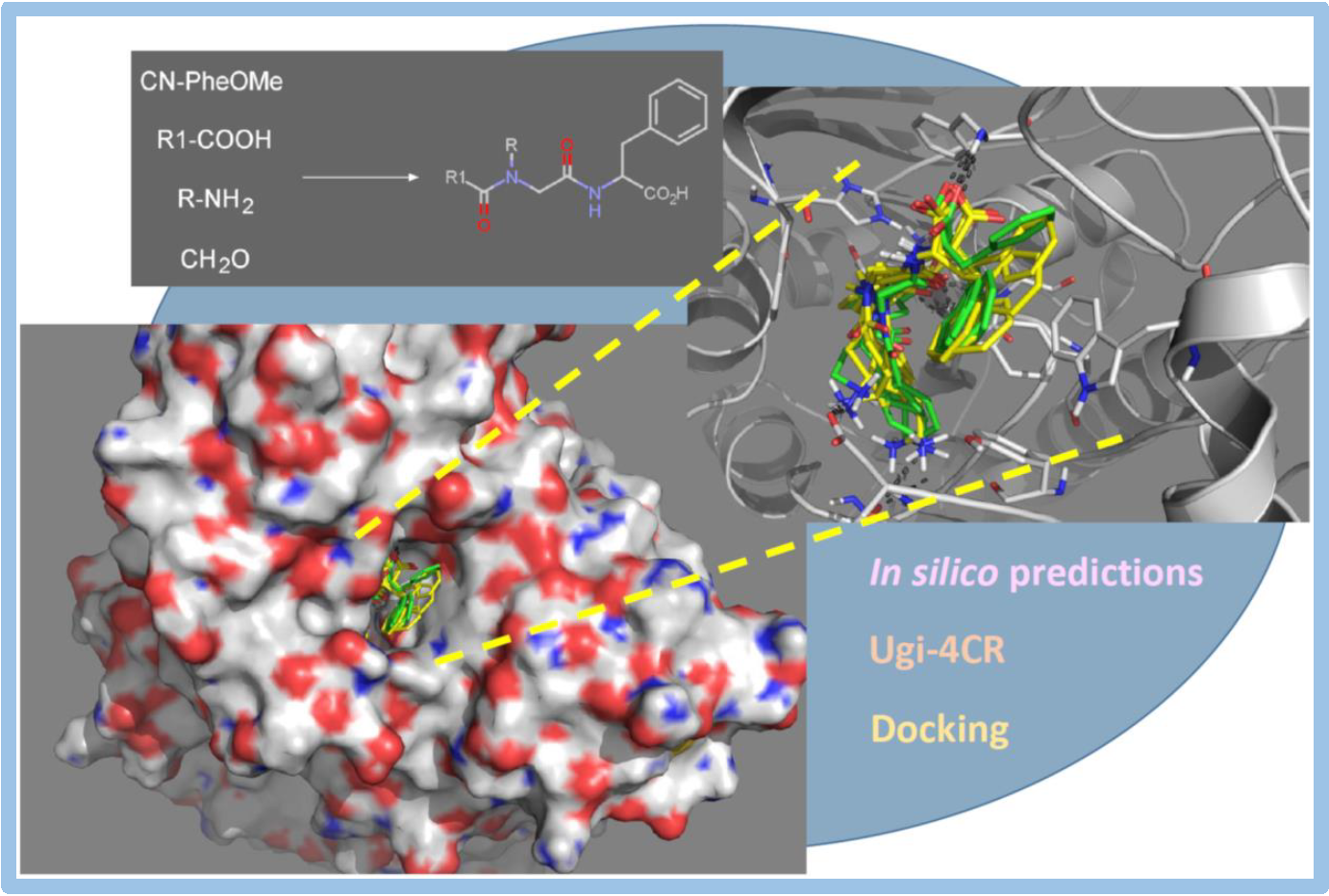
121.
J.L. Velázquez-Libera, J. Caballero*, A.P. Toropova, A.A. Toropov*.
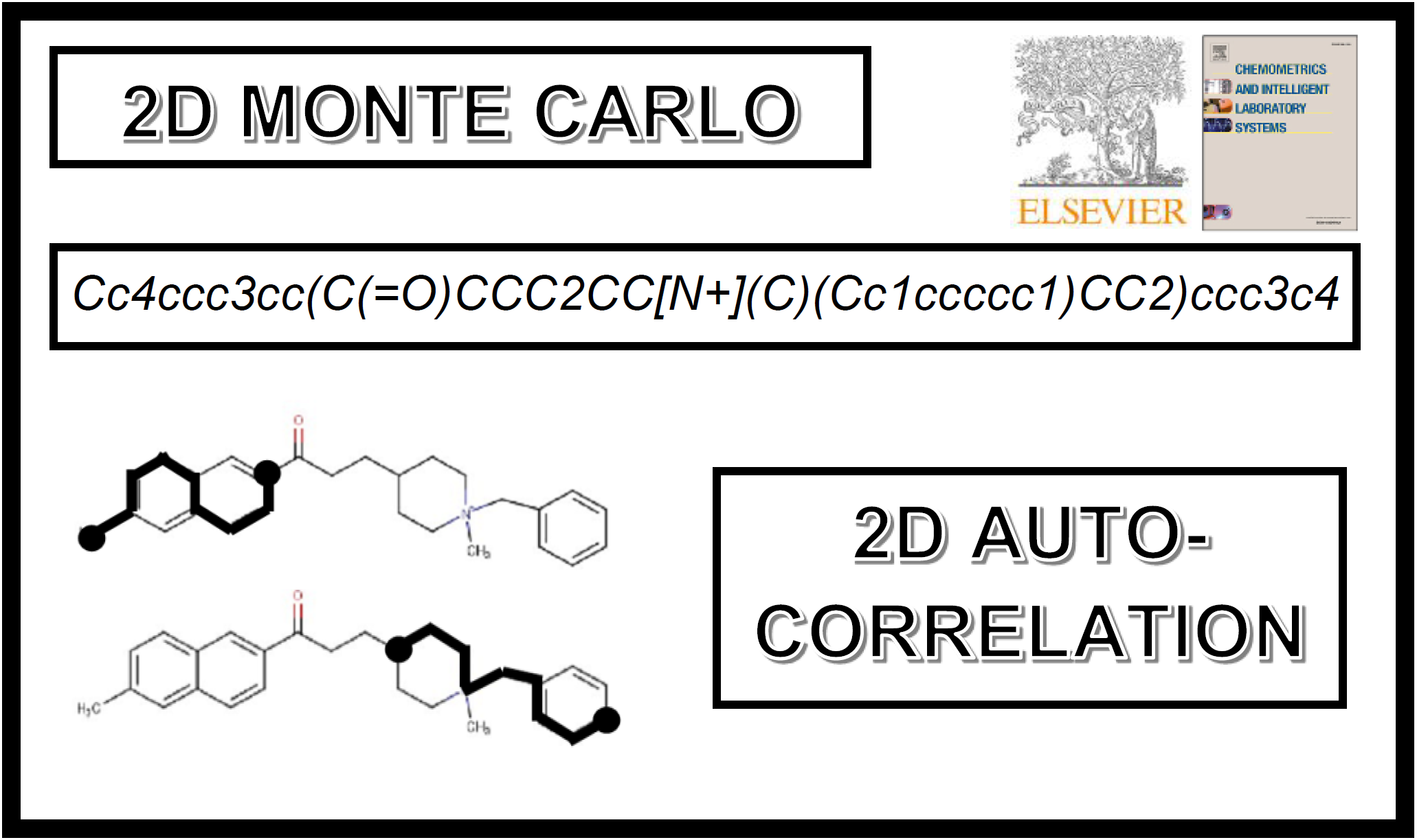
118.
C. Navarro-Retamal, A. Bremer, H.I. Ingólfsson, J. Alzate-Morales, J. Caballero, A. Thalhammer, W. González, D.K. Hincha
116.
K. Mena-Ulecia, F. Gonzalez-Norambuena, A. Vergara-Jaque, H. Poblete, W. Tiznado, J. Caballero*
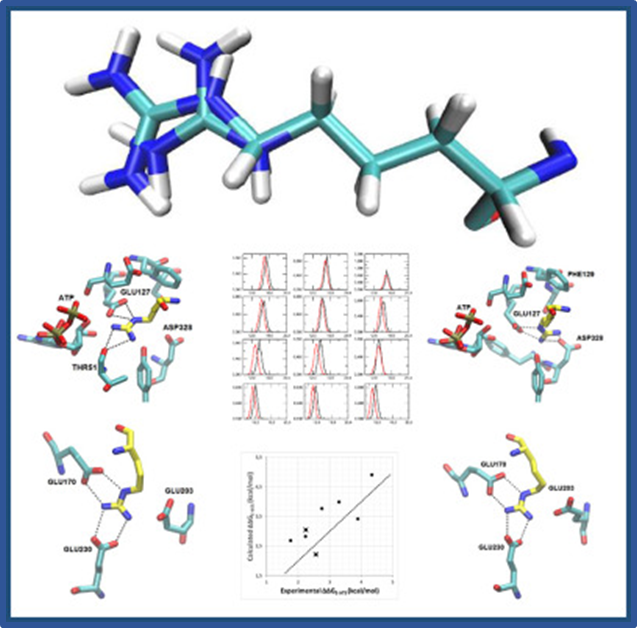
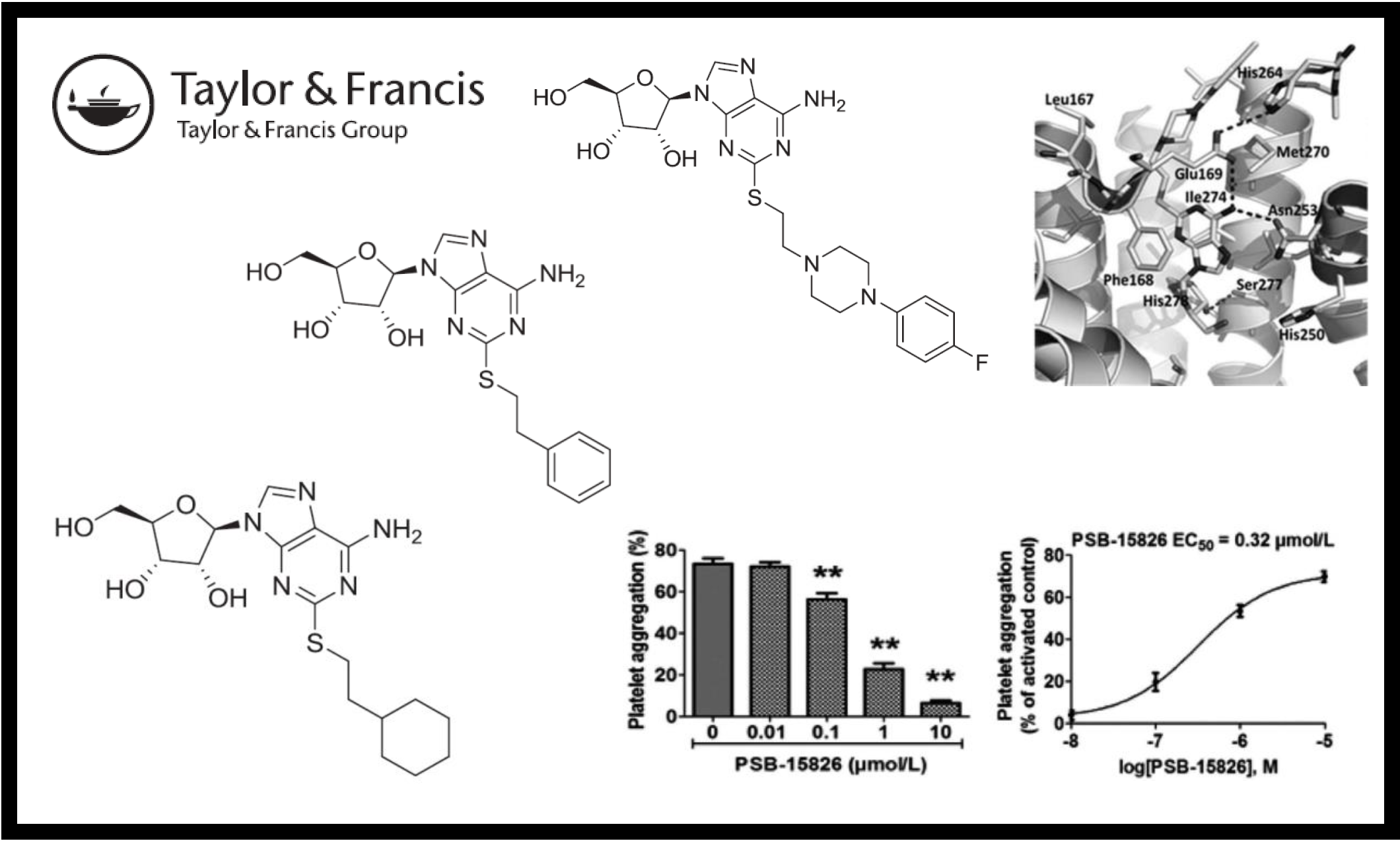
114.
E. Fuentes*, M. Fuentes, J. Caballero, I. Palomo, S. Hinz, A. El-Tayeb, C.E. Müller*
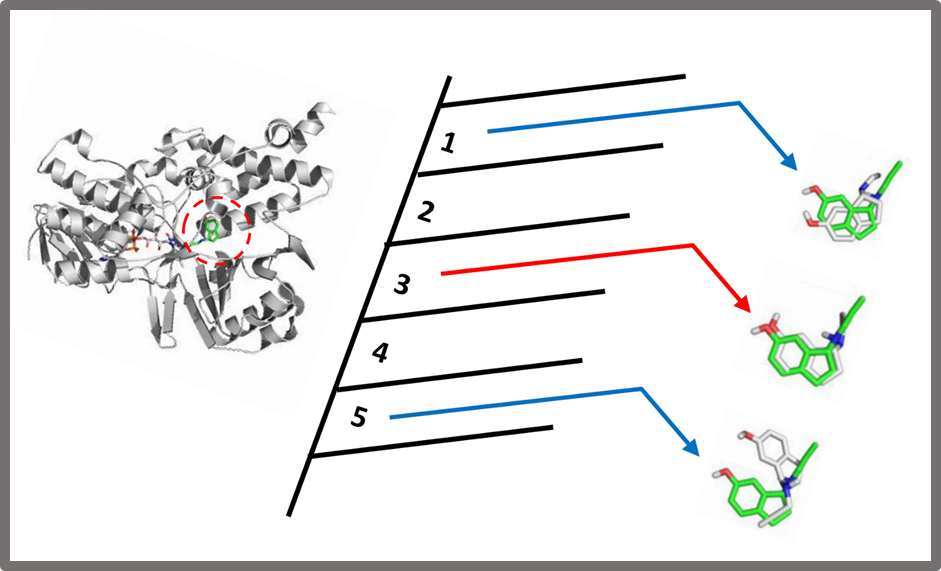
113.
P. Pallavi, M. Pretze, J. Caballero, Y. Li, B.B. Hofmann, E. Stamellou, S. Klotz, C. Wängler, B. Wängler, R. Loesel, S. Roth, B. Theisinger, H. Moerz, U. Binzen, W. Greffrath, R.D. Treede, M.C. Harmsen, B.K. Krämer, M. Hafner, B.A. Yard*, A.I. Kälsch
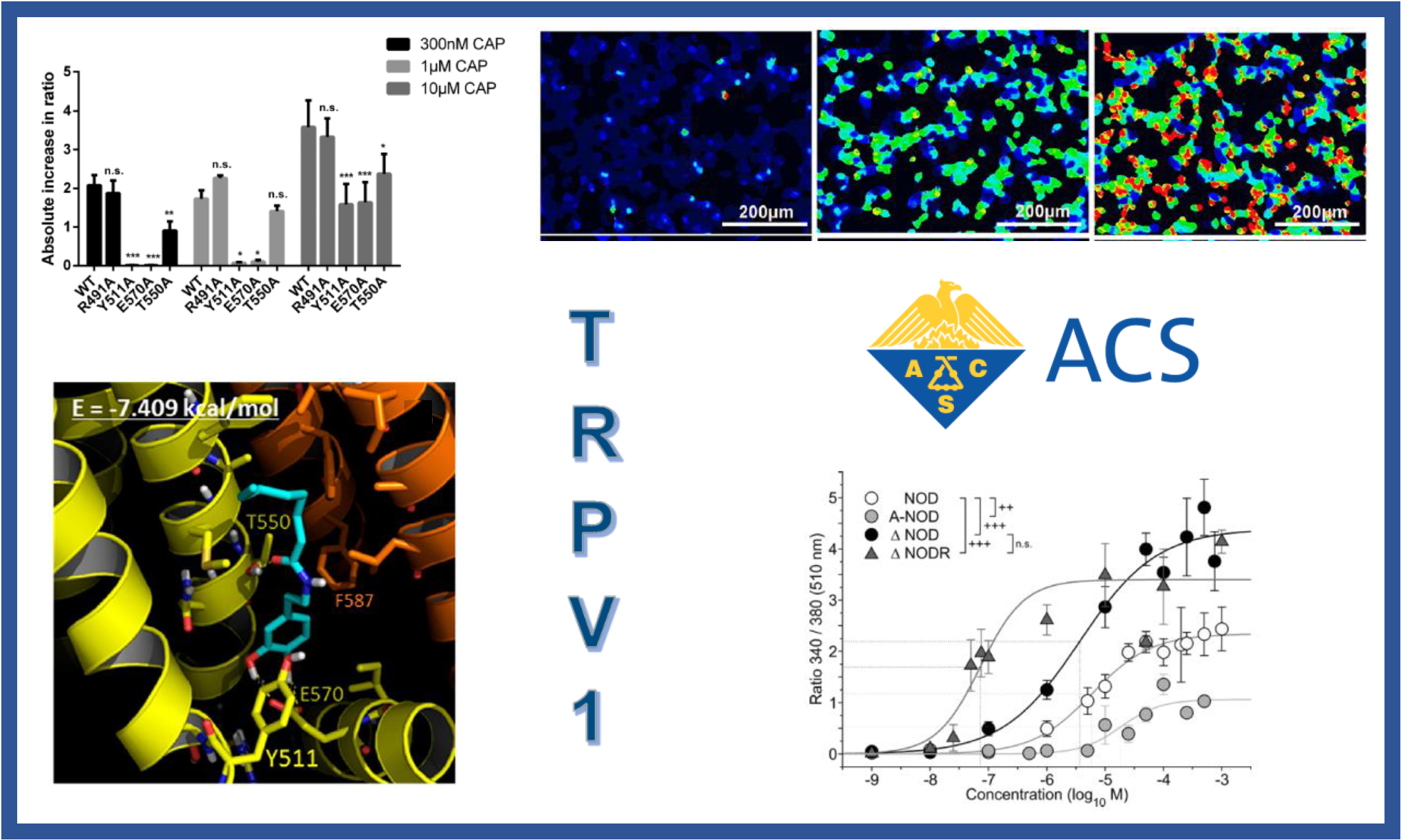
112.
F. Avila-Salas, A. Marican, J. Villaseñor, M. Arenas-Salinas, Y. Argandoña, J. Caballero*, E.F. Durán-Lara*
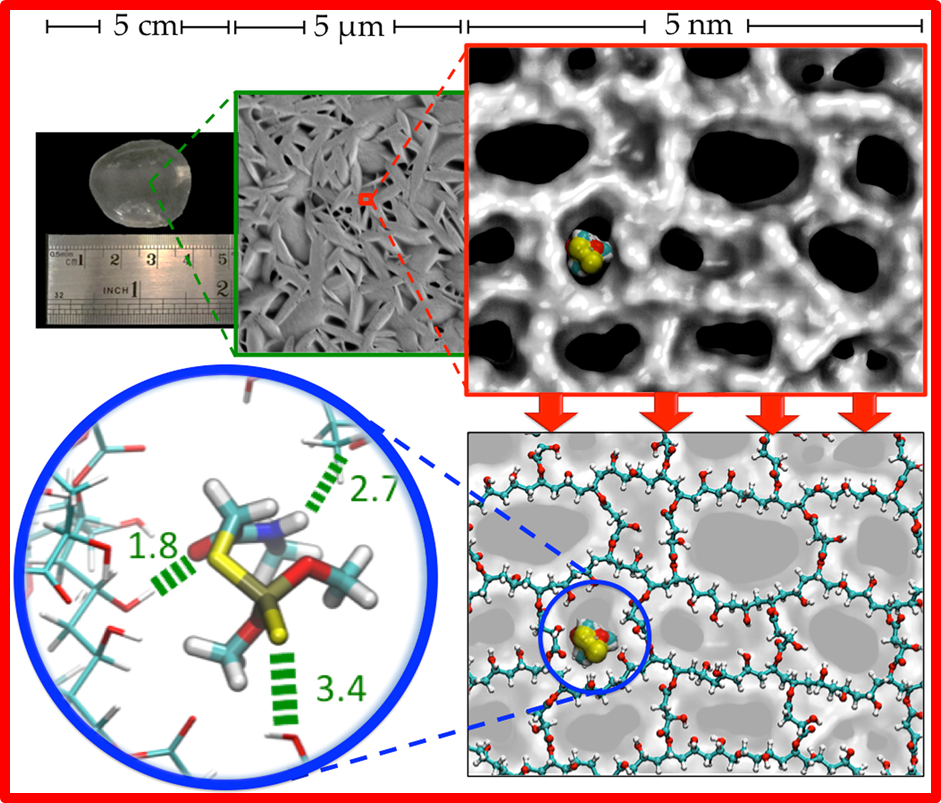
111.
P. Ojeda*, D. Ramírez, J. Alzate-Morales, J. Caballero, Q. Kaas, W. González*
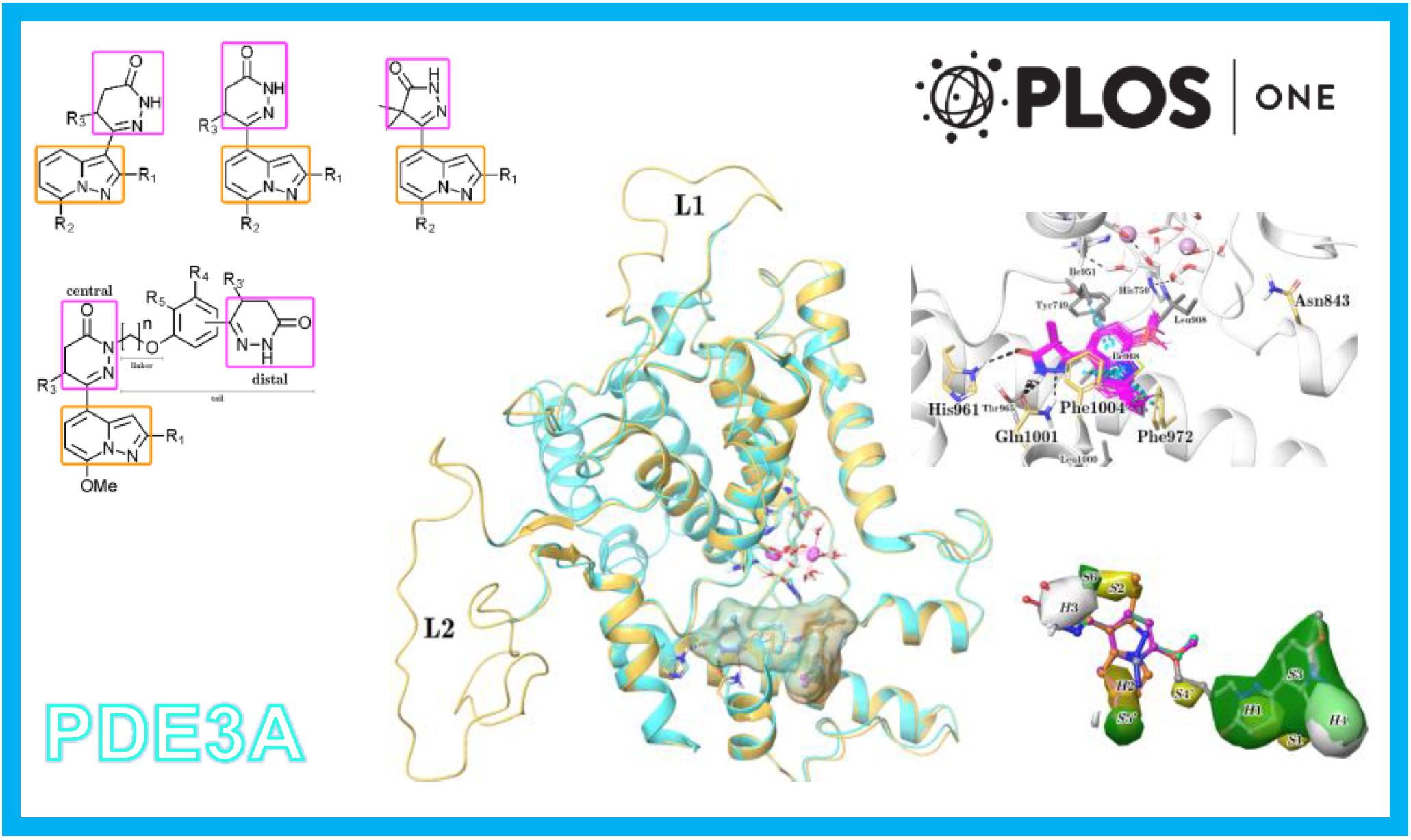
110.
C. Muñoz-Gutierrez, D. Caceres-Rojas, F. Adasme-Carreño, I. Palomo, E. Fuentes, J. Caballero*.
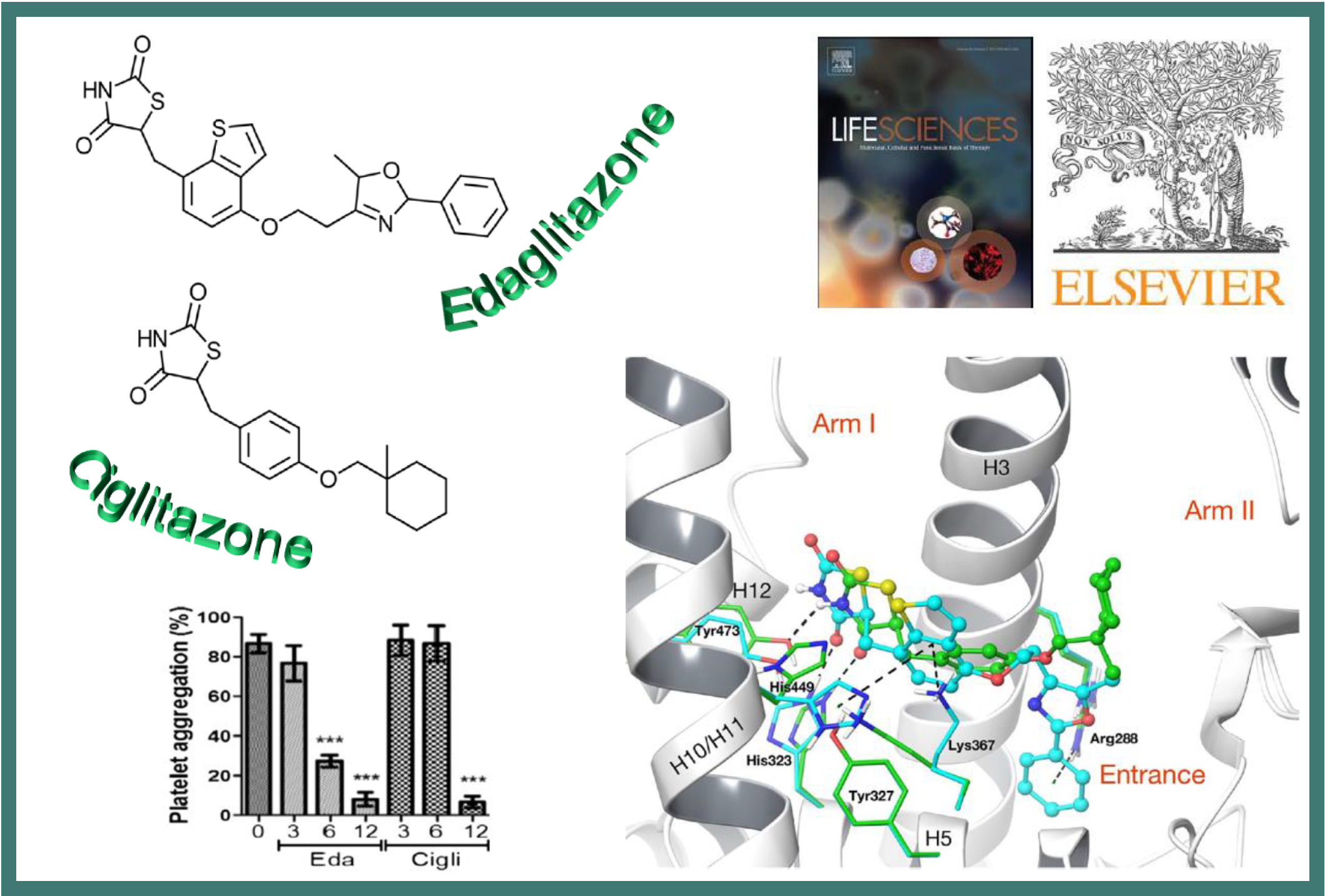
109.
C. Muñoz-Gutierrez, C. Sepulveda, J. Caballero*, I. Palomo, E. Fuentes*
108.
A. Cornejo*, F.A. Sandoval, L. Caballero, L. Machuca, P. Muñoz, J. Caballero, G. Perry, A. Ardiles, C. Areche, F. Melo
107.
K. Sklodowski, J. Riedelsberger*, N. Raddatz, G. Riadi, J. Caballero, I. Chérel, W. Schulze, A. Graf, I. Dreyer*
105.
R.K. Sharma, M. Espinoza-Moraga, H. Poblete, R.G. Douglas, E.D. Sturrock, J. Caballero*, K. Chibale*
104.
M. Pretze*, P. Pallavi, M. Roscher, S. Klotz, J. Caballero, U. Binzen, W. Greffrath, R.D. Treede, M. Harmsen, M. Hafner, B. Yard, C. Wängler, B. Wängler*
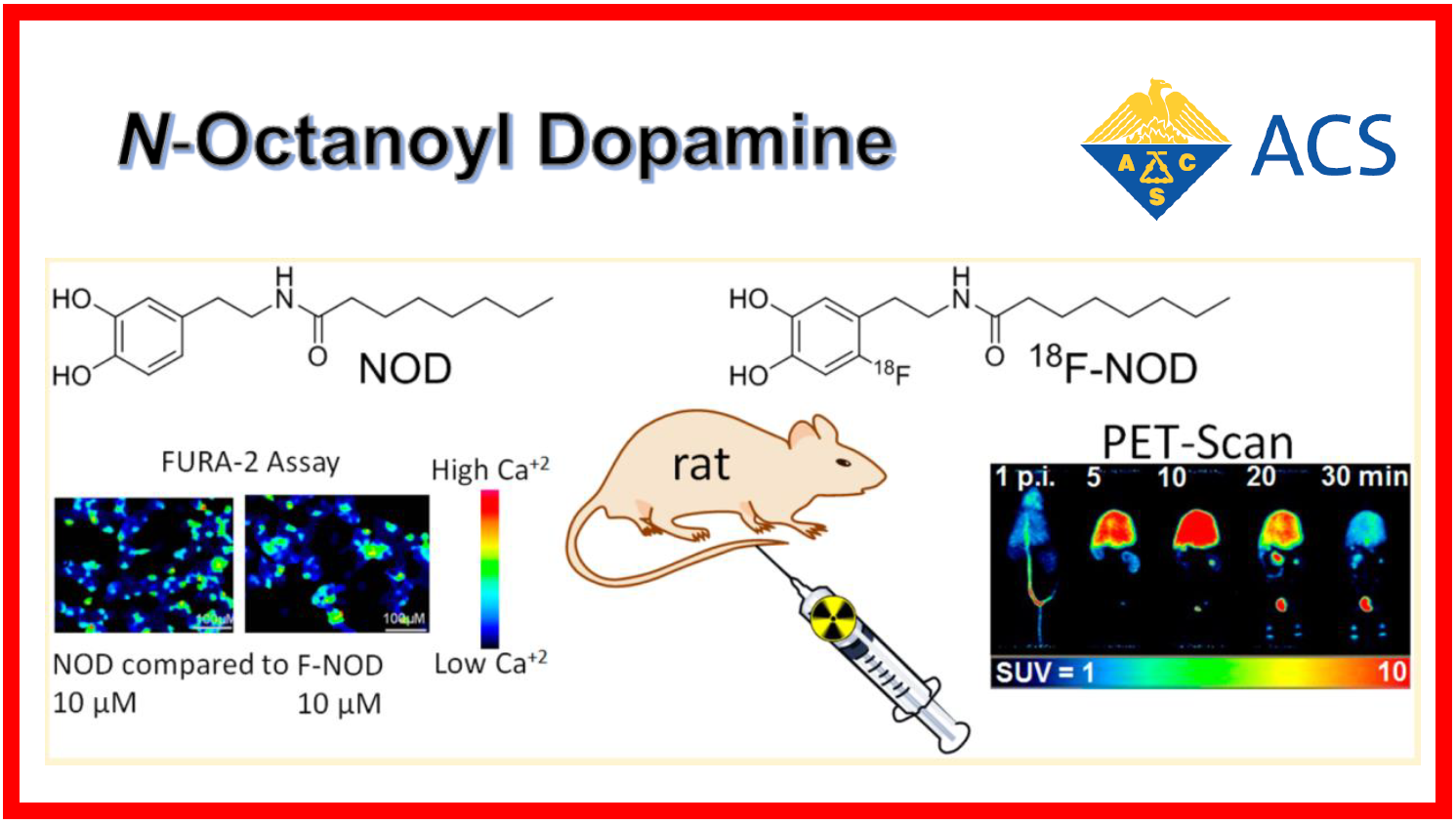
103.
C. Navarro-Retamal, A. Bremer, J. Alzate-Morales, J. Caballero, D.K. Hincha, W. Gonzalez*, A. Thalhammer*
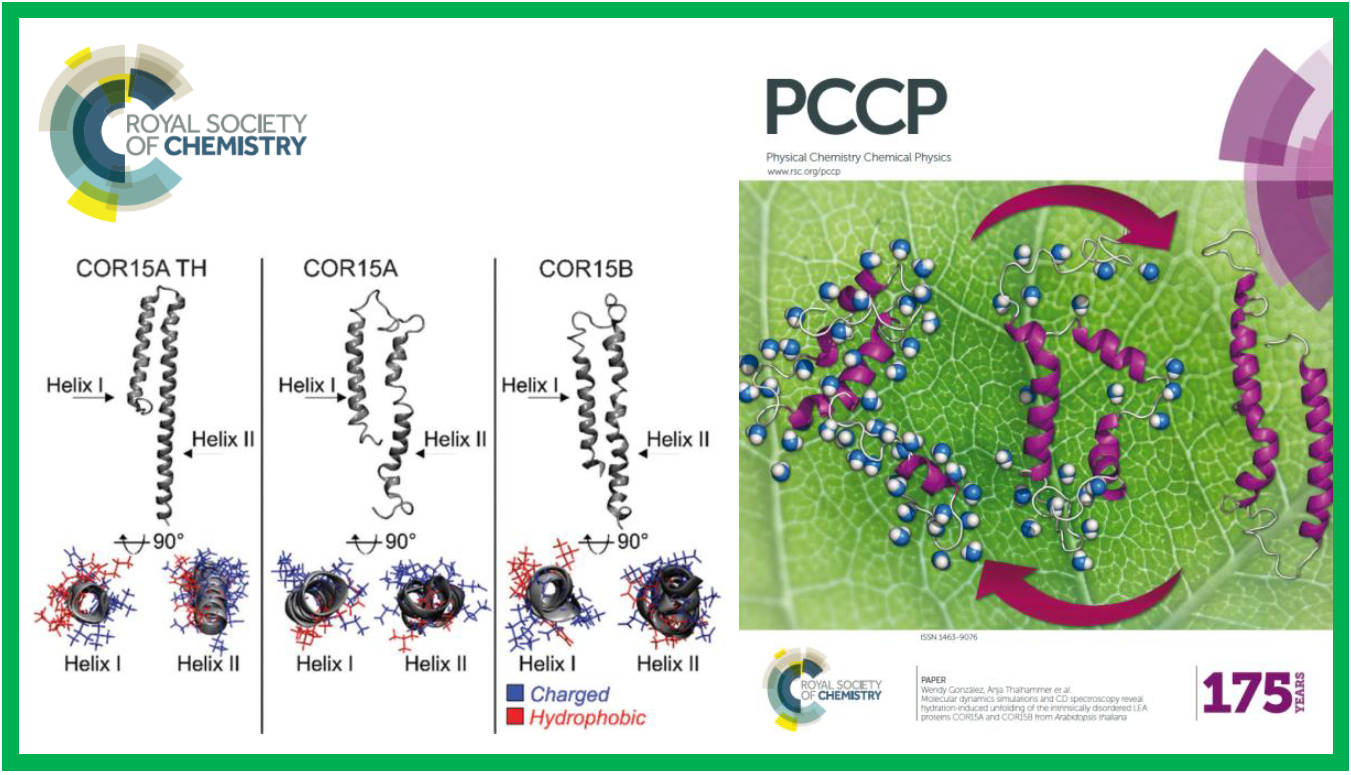
102.
F. Reissig*, C. Mamat, J. Steinbach, H.-J. Pietzsch, R. Freudenberg, C. Navarro-Retamal, J. Caballero, J. Kotzerke, G. Wunderlich*
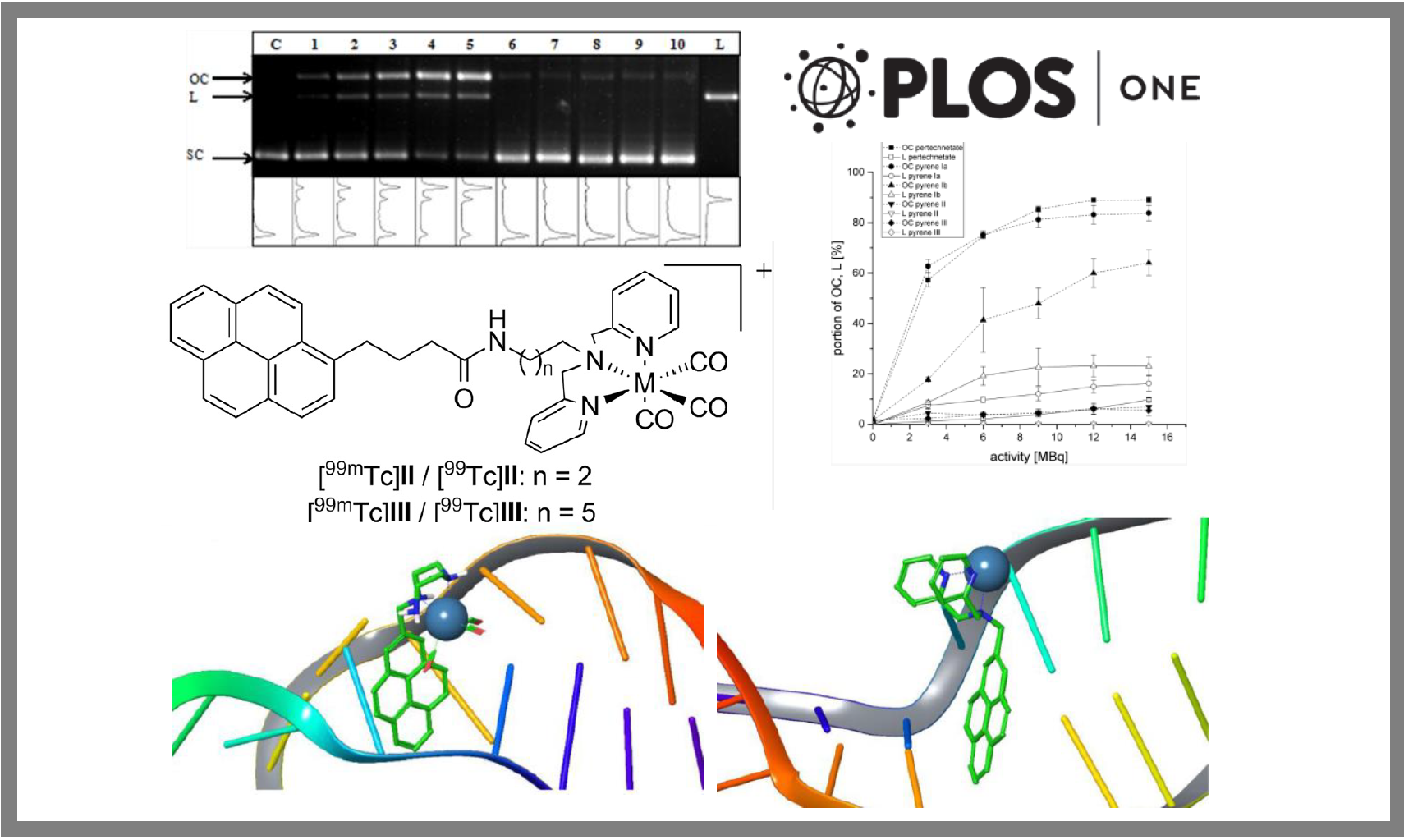
101.
A. Cornejo*, F. Salgado, J. Caballero, R. Vargas, M. Simirgiotis, C. Areche*
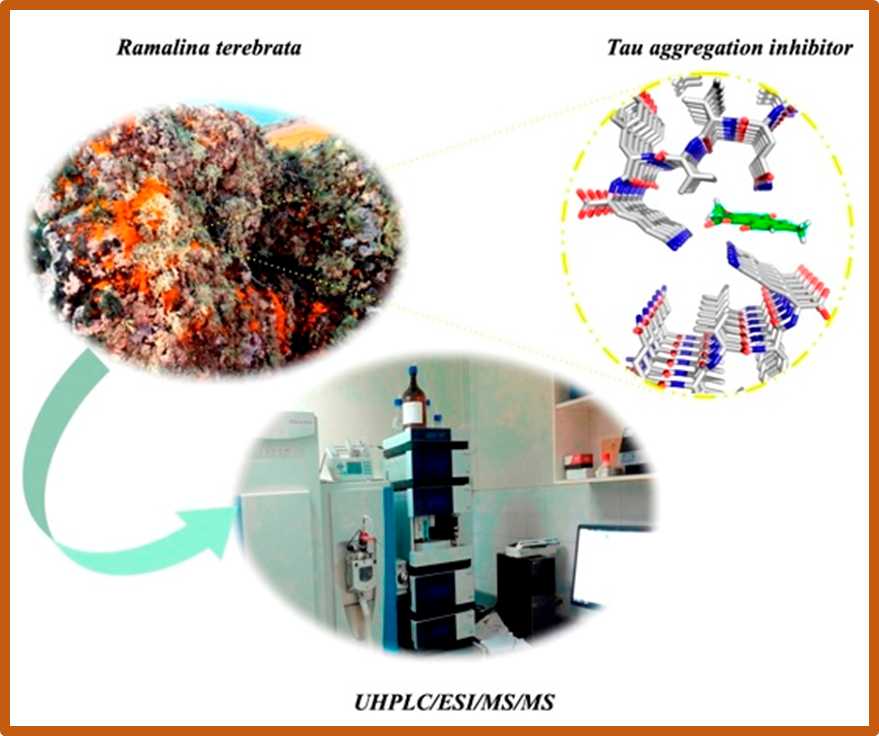
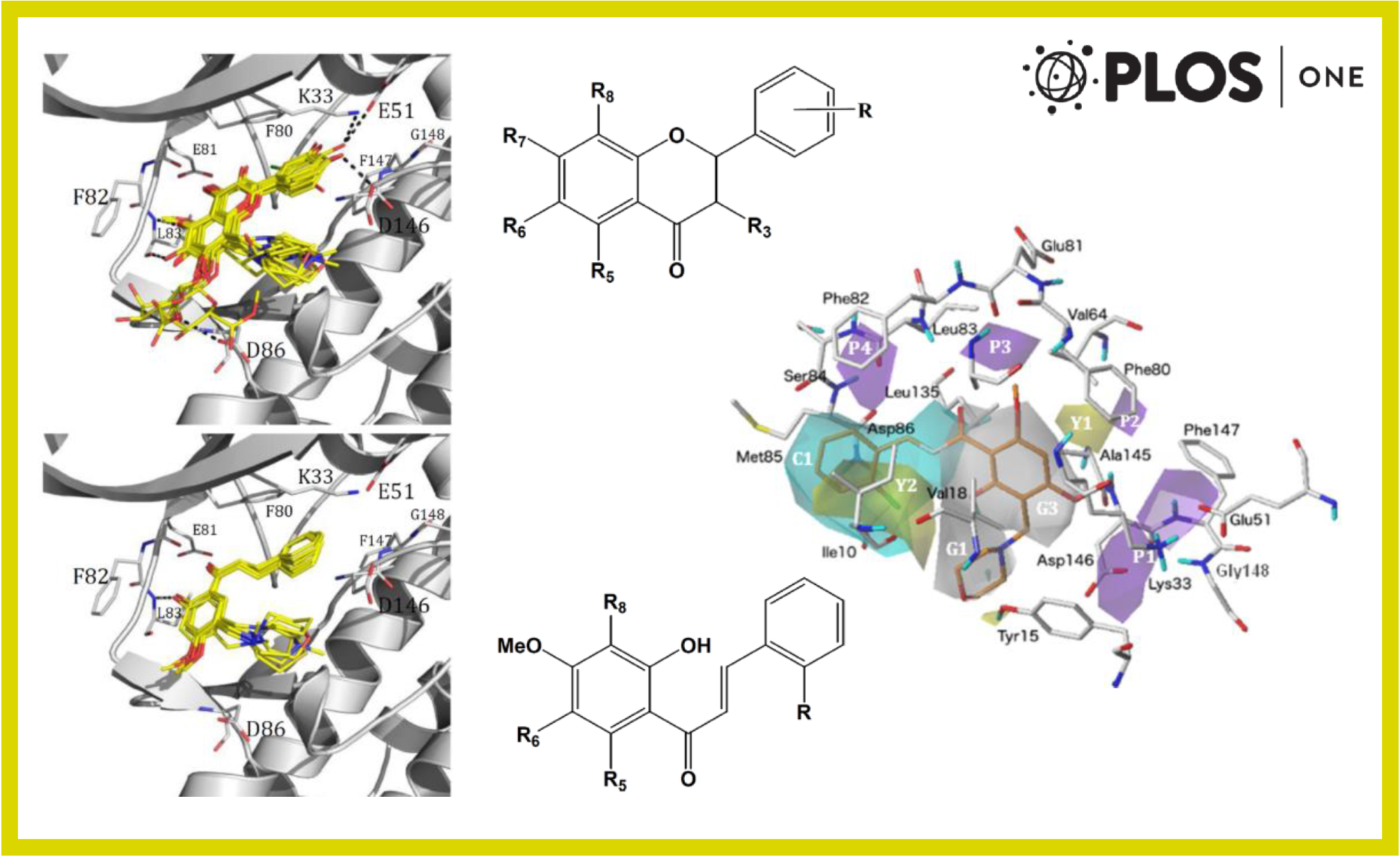
99.
C. Muñoz-Gutierrez, F. Adasme-Carreño, E. Fuentes, I. Palomo, J. Caballero*
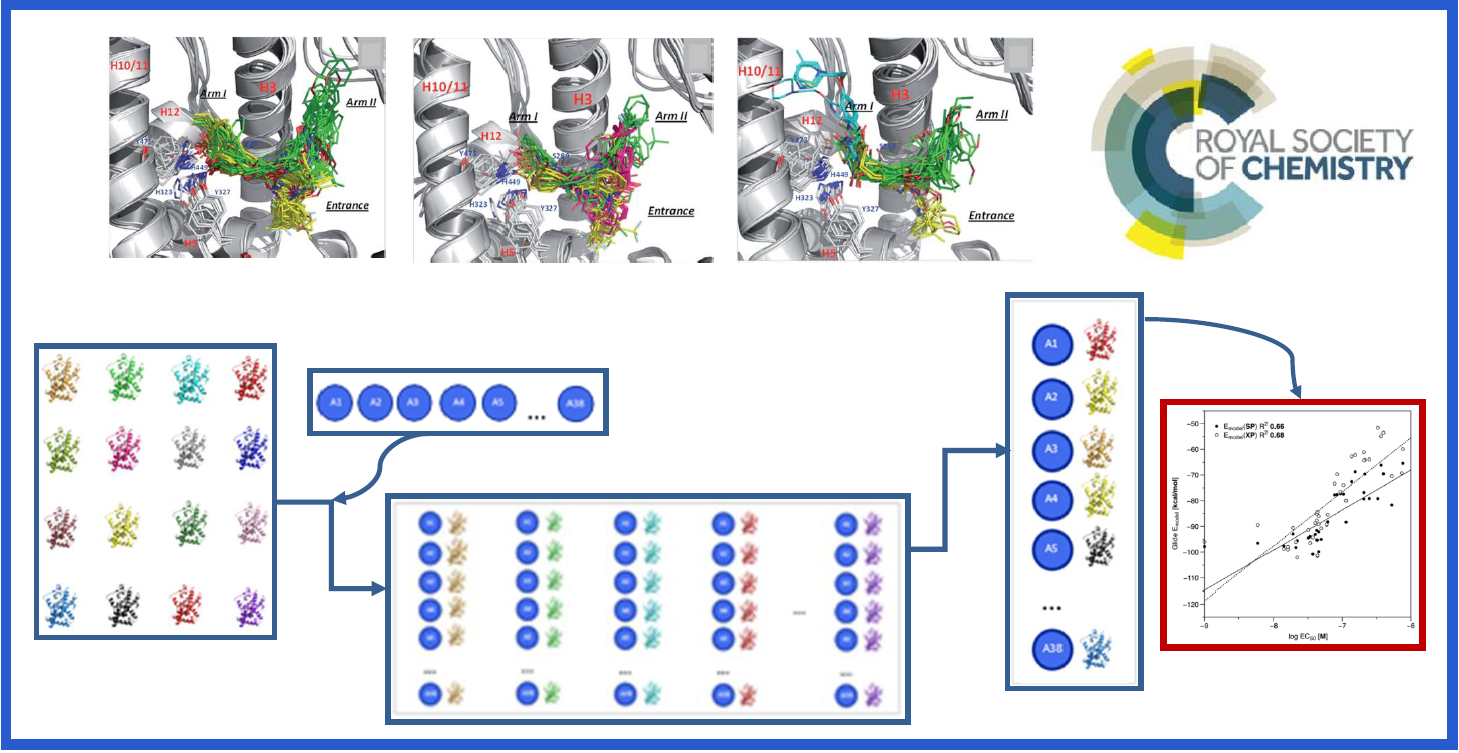
96.
C. Navarro-Retamal, C. Gaete-Eastman, R. Herrera*, J. Caballero, J.H. Alzate-Morales*
95.
P. De-la-Torre, M. Gutierrez*, J. Caballero, J. Trilleras, L. Astudillo, A. Cardenas, I. Brito
94.
P. De-la-Torre*, A.V. Treuer, M. Gutiérrez, H. Poblete, J.H. Alzate-Morales, J. Trilleras, L. Astudillo-Saavedra, J. Caballero*
91.
K. Ebert, J. Wiemer, J. Caballero, M. Köckerling, J. Steinbach, J. Pietzsch, C. Mamat*
89.
W. González*, B. Valdebenito, J. Caballero, G. Riadi, J. Riedelsberger, G. Martínez, D. Ramírez, L. Zúñiga, F.V. Sepúlveda, I. Dreyer, M. Janta, D. Becker*
87.
L. Quesada-Romero, K. Mena-Ulecia, M. Zuñiga, P. De-la-Torre, D. Rossi, W. Tiznado, S. Collina, J. Caballero*
86.
E. Fuentes, J. Pereira, D. Mezzano, M. Alarcón, J. Caballero, P. Pérez, I. Palomo*
85.
K. Mena-Ulecia, A. Vergara-Jaque, H. Poblete, W. Tiznado, J. Caballero*
83.
F. Adasme-Carreño, C. Muñoz-Gutierrez, J. Caballero, J.H. Alzate-Morales*
82.
P. De-la-Torre*, E. Osorio, J.H. Alzate-Morales, J. Caballero, J. Trilleras, L. Astudillo-Saavedra, I. Brito, A. Cárdenas, J. Quiroga, M. Gutiérrez*
81.
J. Torres-Vega, A. Vasquez-Espinal, J. Caballero, M. Valenzuela, L. Alvarez-Thon, E. Osorio, W. Tiznado*
79.
E. Fuentes, L. Badimon, J. Caballero, T. Padró, G. Vilahur, M. Alarcón, P. Pérez, I. Palomo*
78.
O. García-Beltrán*, O. Yañez, J. Caballero, A. Galdámez, N. Mena, M.T. Nuñez, B.K. Cassels
75.
O. García-Beltrán*, B.K. Cassels, N. Mena, M.T. Nuñez, O. Yañez, J. Caballero
74.
O. García-Beltrán*, N. Mena, O. Yañez, J. Caballero, V. Vargas, M.T. Nuñez, B.K. Cassels
70.
J. Caballero, C. Muñoz, J.H. Alzate-Morales, S. Cunha, L. Gano, R. Bergmann, J. Steinbach, T. Kniess*
69.
P. De La Torre, L. Astudillo Saavedra, J. Caballero, J. Quiroga, J.H. Alzate-Morales, M. Gutiérrez Cabrera*, J. Trilleras*
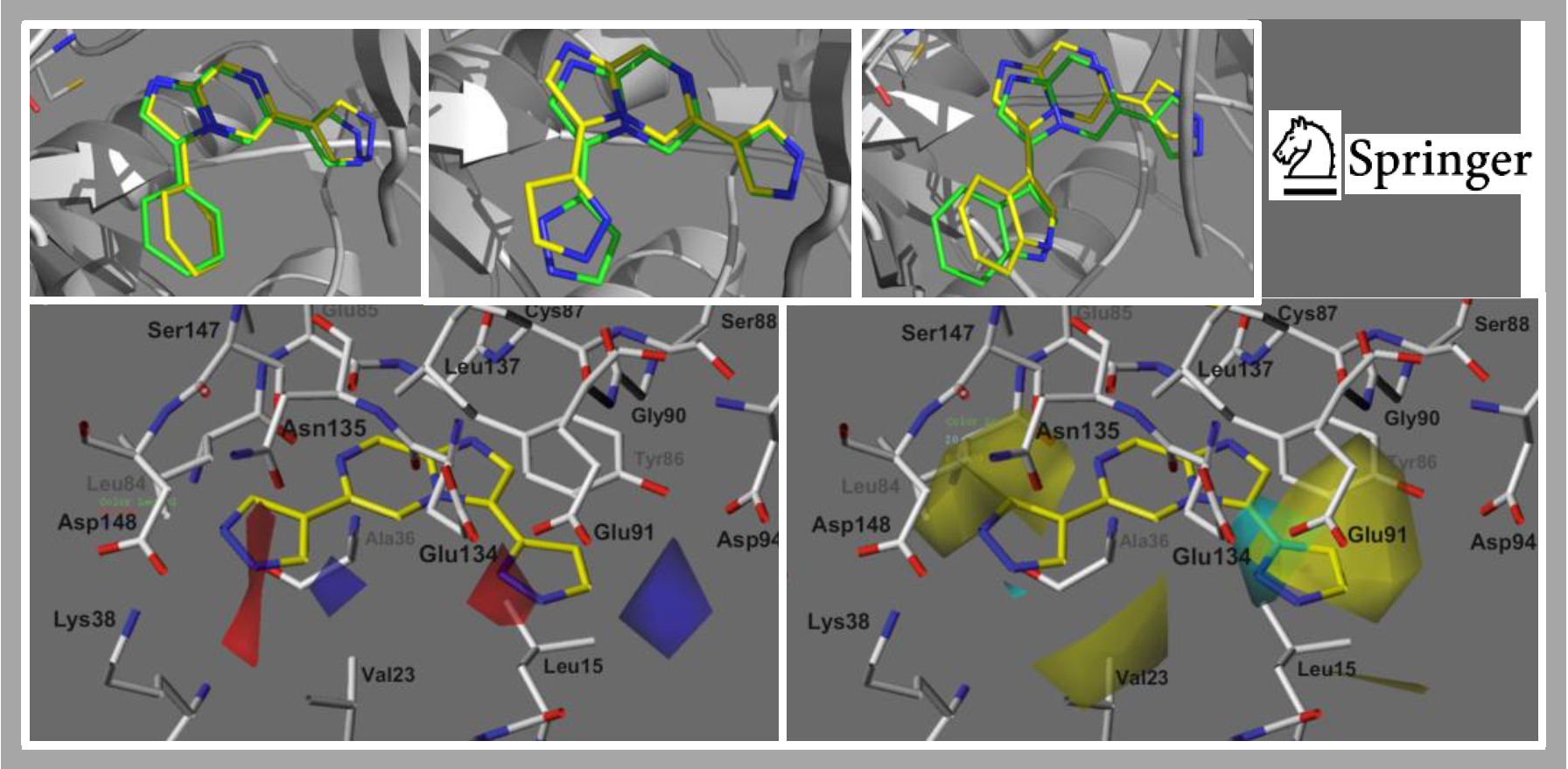
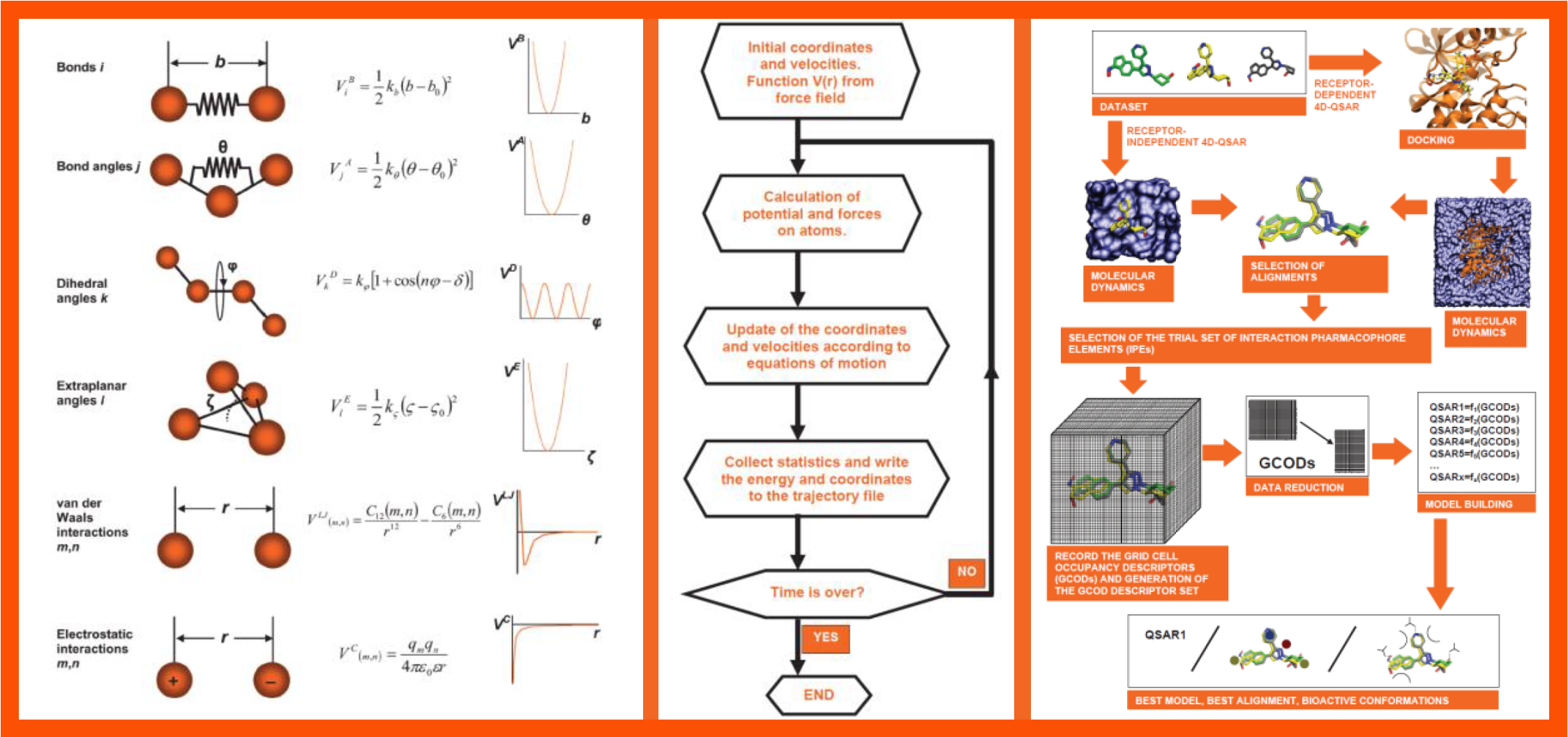
66.
J.Z. Chandanshive, P.B. González, W. Tiznado, B.F. Bonini, J. Caballero, C. Femoni, M. Comes Franchini*
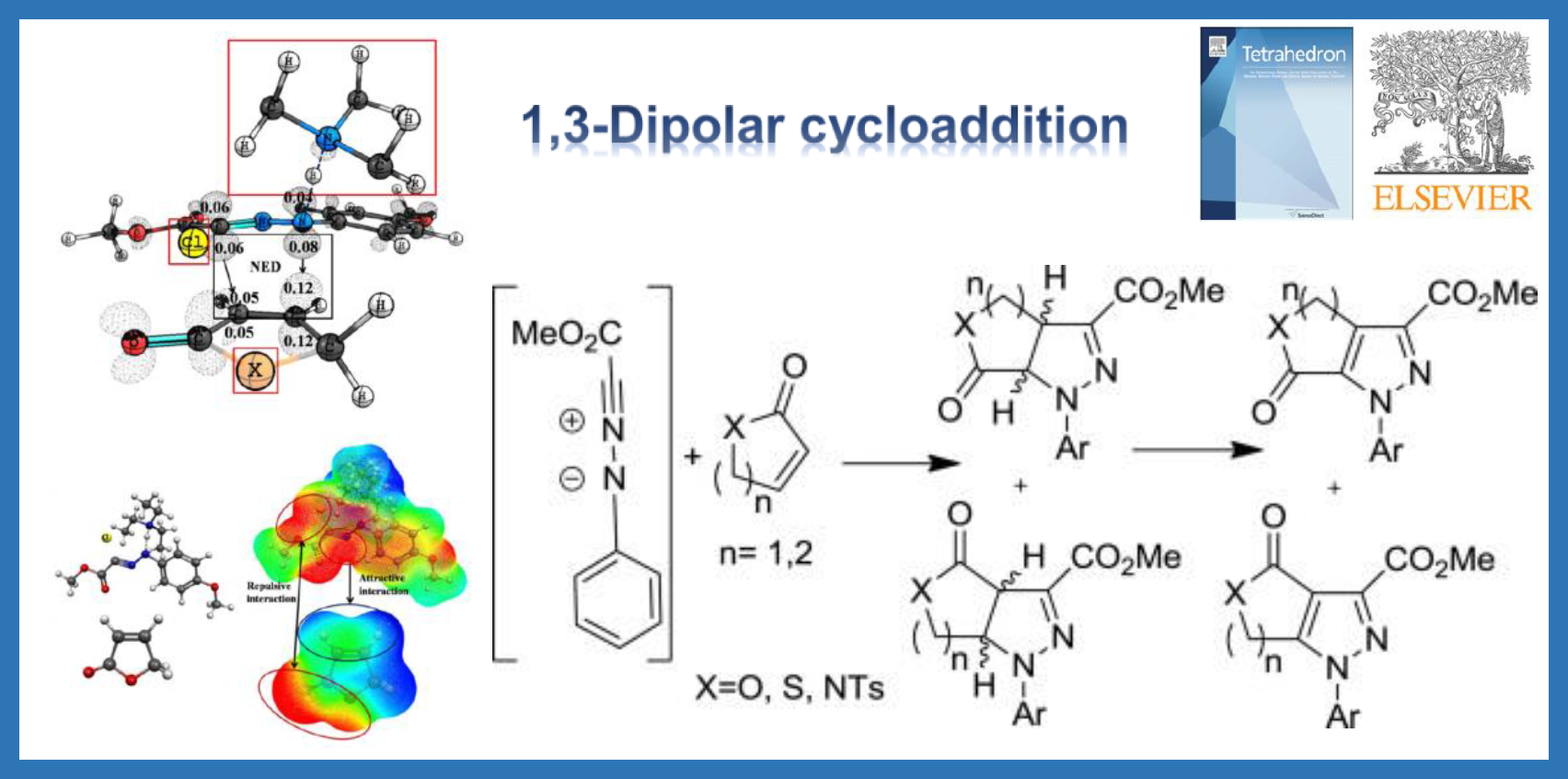
65.
M. Espinoza-Moraga, J. Caballero, F. Gaube, T. Winckler, L.S. Santos*
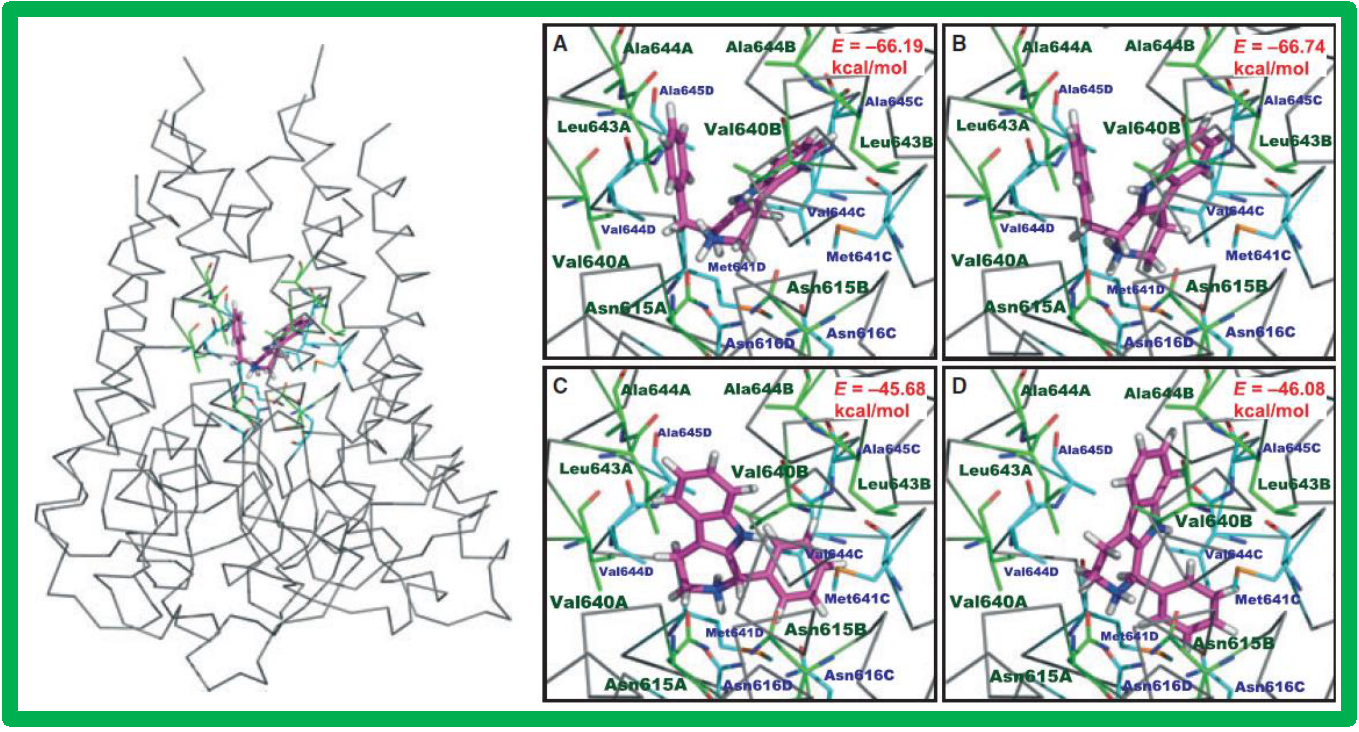
64.
F. Avila-Salas, C. Sandoval, J. Caballero, S. Guiñez-Molinos, L.S. Santos, R.E. Cachau, F.D. González-Nilo*
63.
W. González*, J. Riedelsberger, S.E. Morales-Navarro, J. Caballero, J.H. Alzate-Morales, F.D. González-Nilo, I. Dreyer*
61.
Y. Mirabal-Gallardo, M.D.P.C. Soriano, J. Caballero, J.H. Alzate-Morales, M.J. Simirgiotis, L.S. Santos*
60.
C. Muñoz, F. Adasme, J.H. Alzate-Morales, A. Vergara-Jaque, T. Kniess, J. Caballero*
58.
D. Rossi, A. Pedrali, M. Urbano, R. Gaggeri, M. Serra, L. Fernandez, M. Fernandez, J. Caballero, S. Ronsisvalle, O. Prezzavento, D. Schepmann, B. Wünsch, D. Curti, O.Azzolina, S. Collina*
57.
J. Caballero*, S. Zilocchi, W. Tiznado, S. Collina, D. Rossi
56.
J. Z. Chandanshive, B. F. Bonini, W. Tiznado, C. A. Escobar, J. Caballero, C. Femoni, M. Fochi, M. Comes Franchini*
55.
J. Caballero*, M. Quiliano, J.H. Alzate-Morales, M. Zimic, E. Deharo
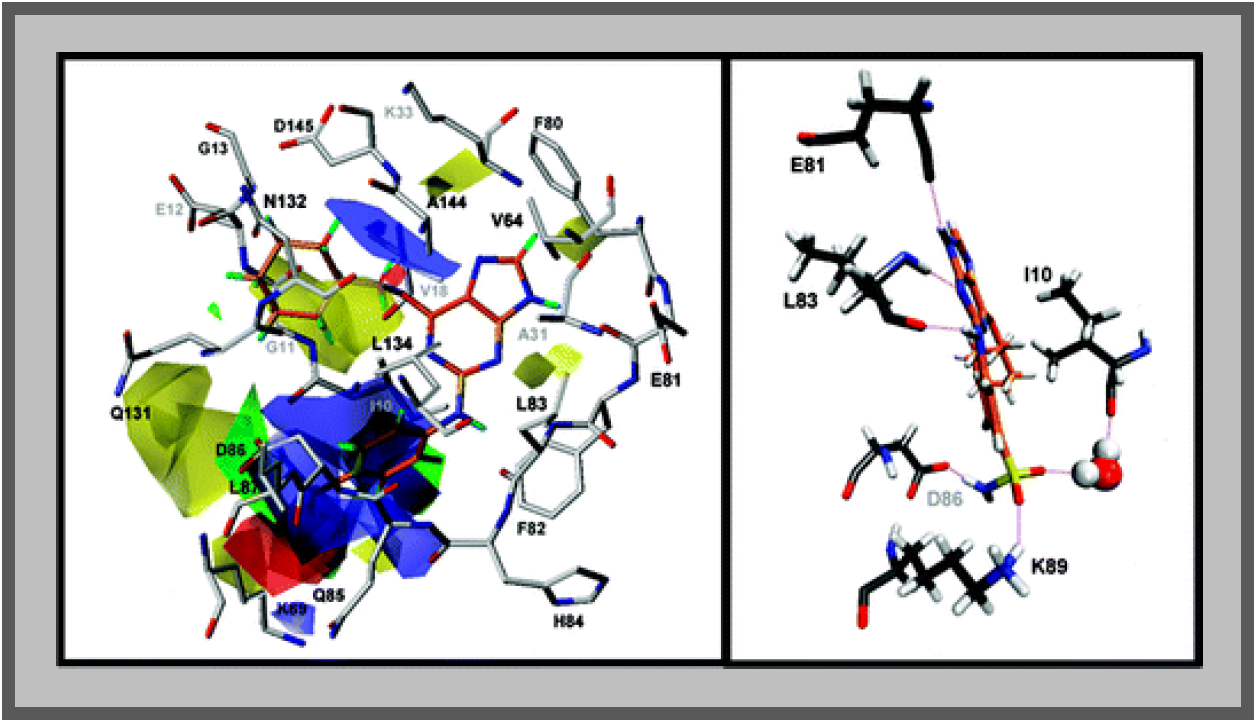
46.
J.H. Alzate-Morales*, J. Caballero, F.D. González-Nilo, R. Contreras
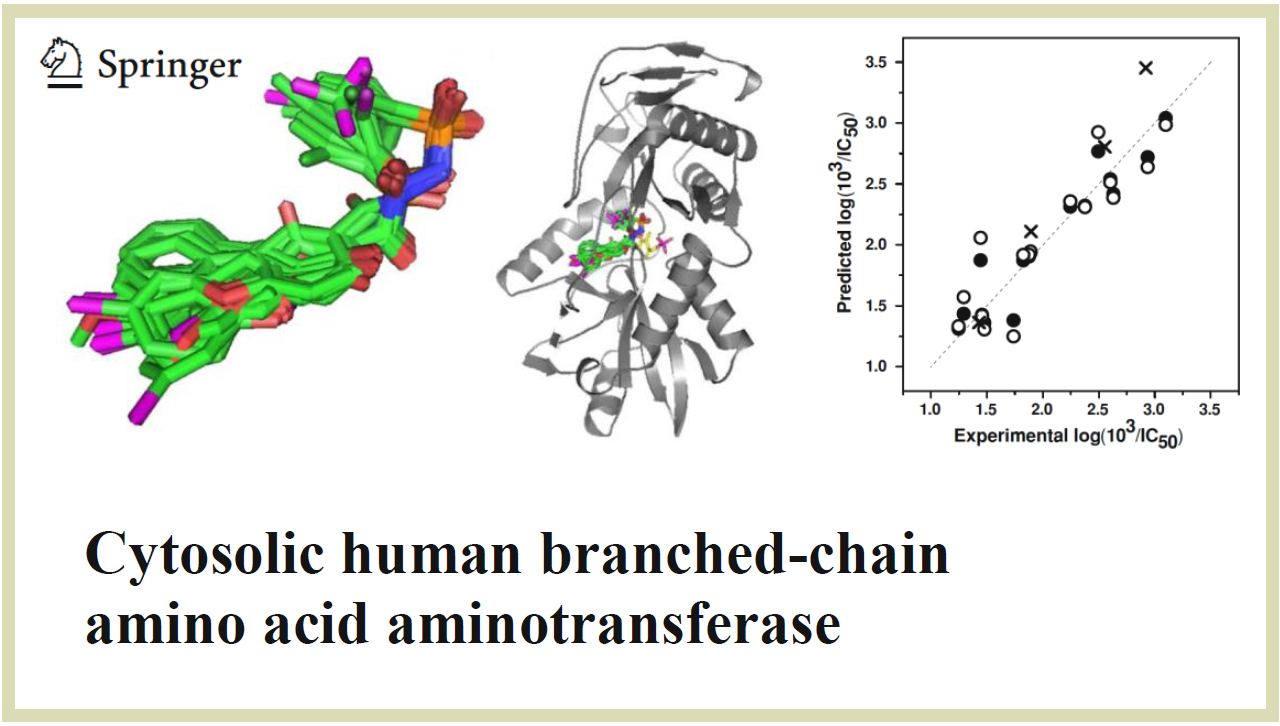
45.
C. Saldías, L. Gargallo*, C. Sandoval, A. Leiva, D. Radic, J. Caballero, M. Saavedra, F.D. González-Nilo
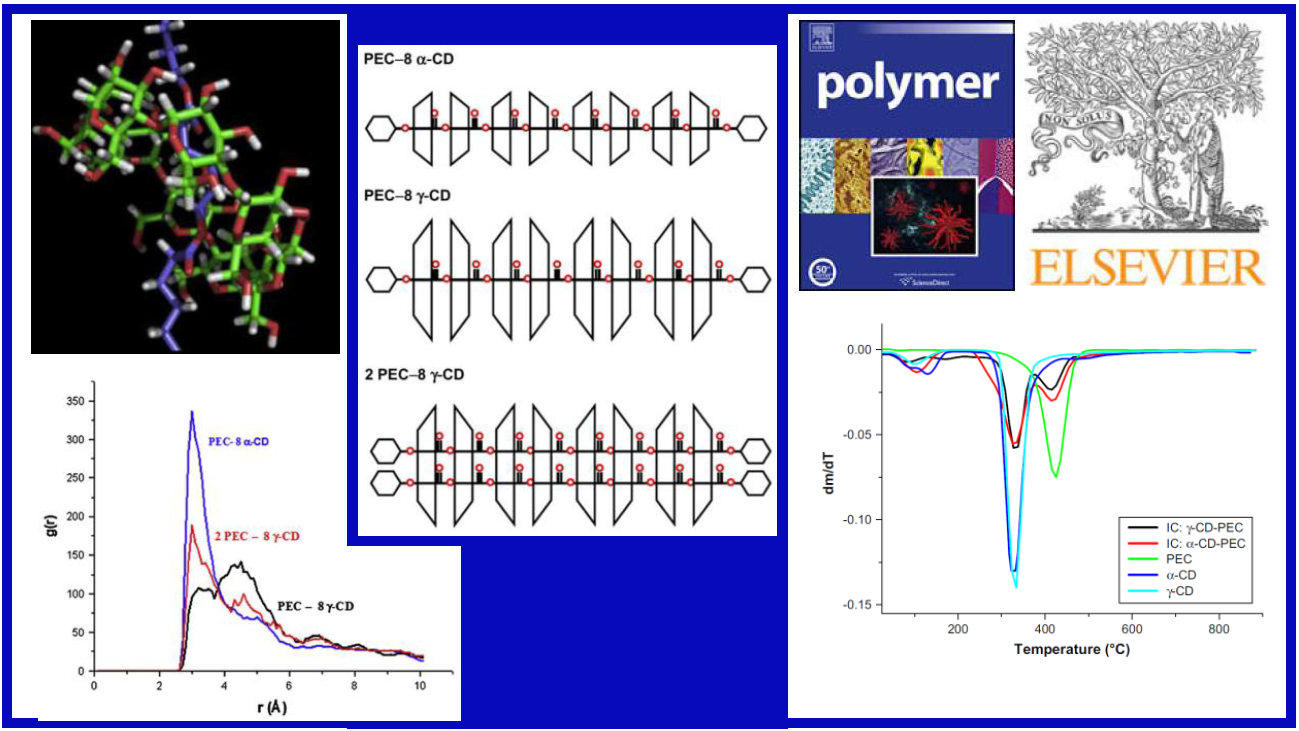
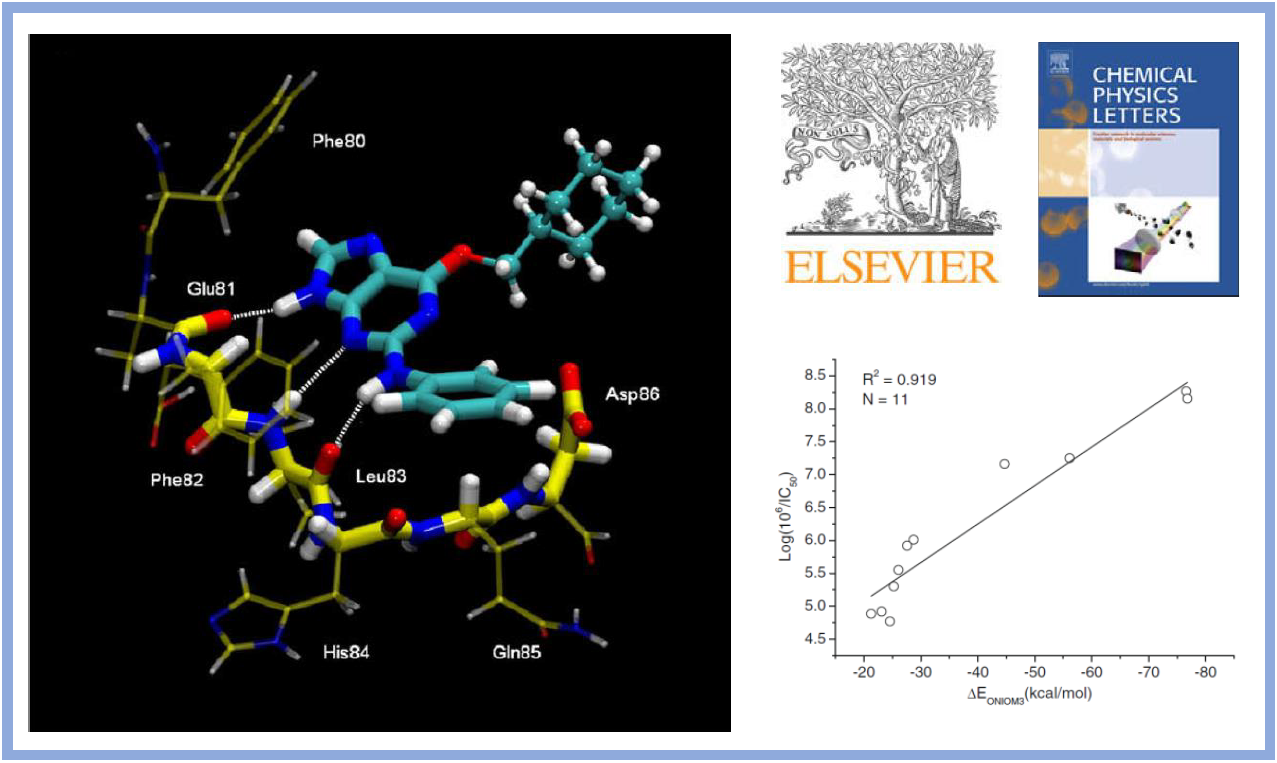
42.
C.F. Lagos*, J. Caballero*, F.D. González-Nilo, C.D. Pessoa-Mahana, T. Pérez-Acle
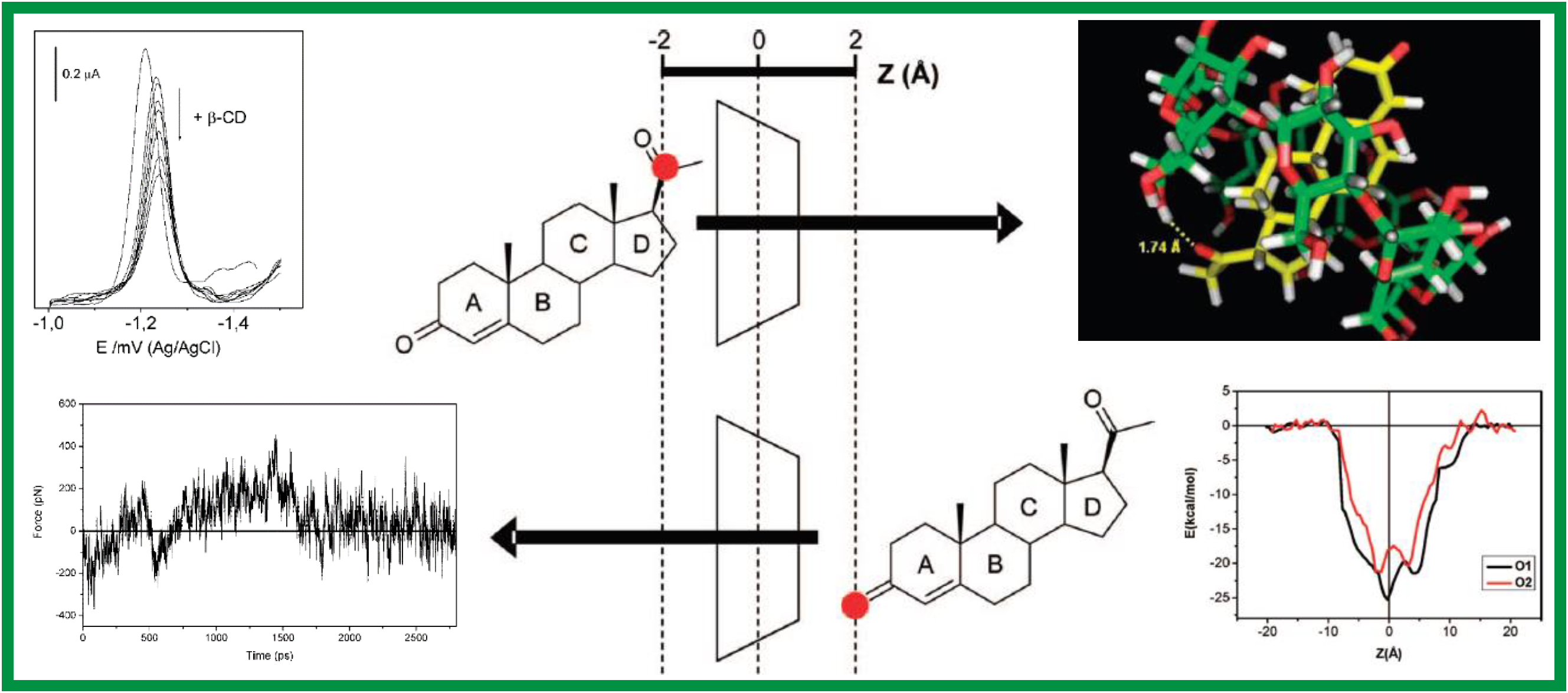
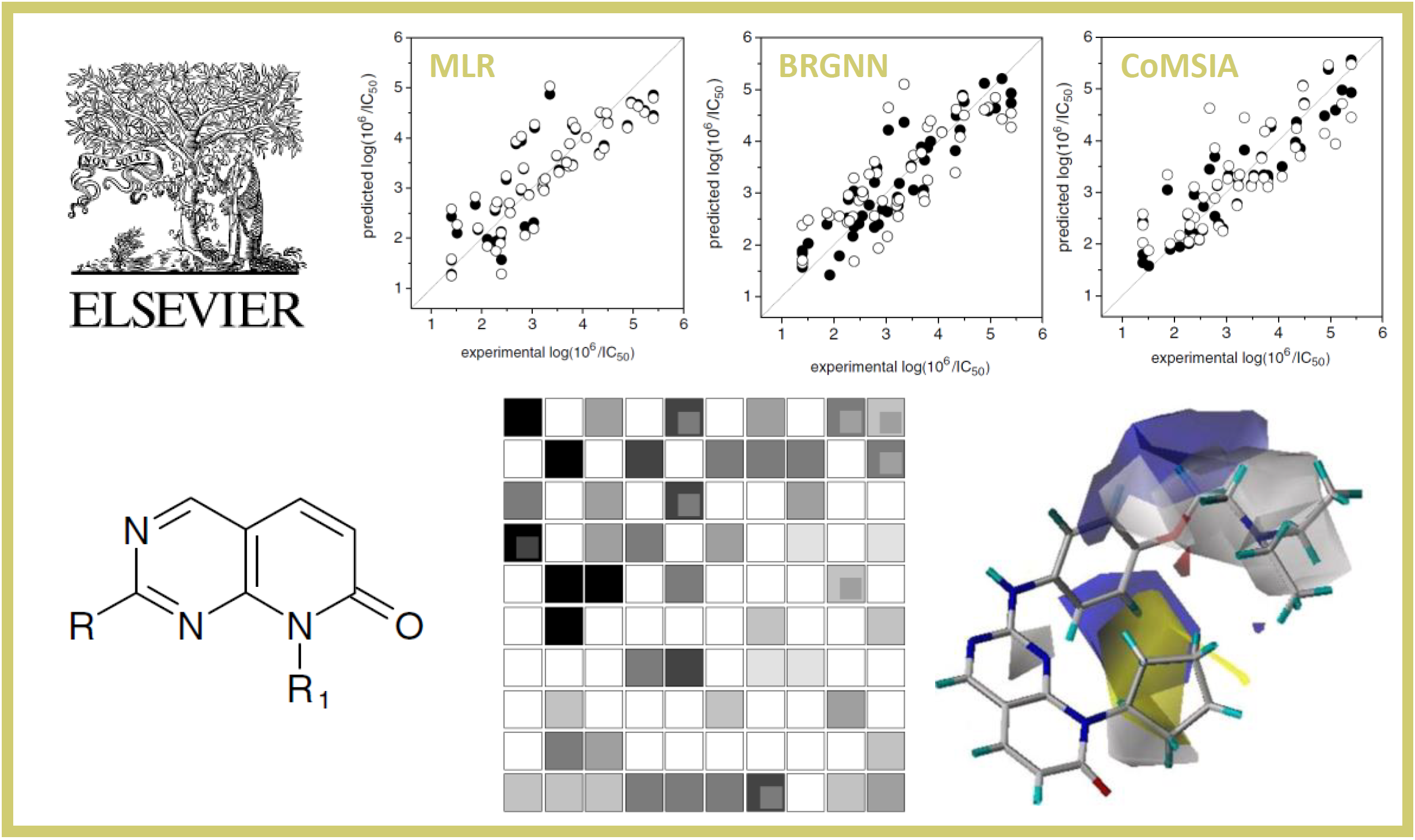
38.
M. Fernández*, L. Fernández, J. Caballero, J.I. Abreu, G. Reyes
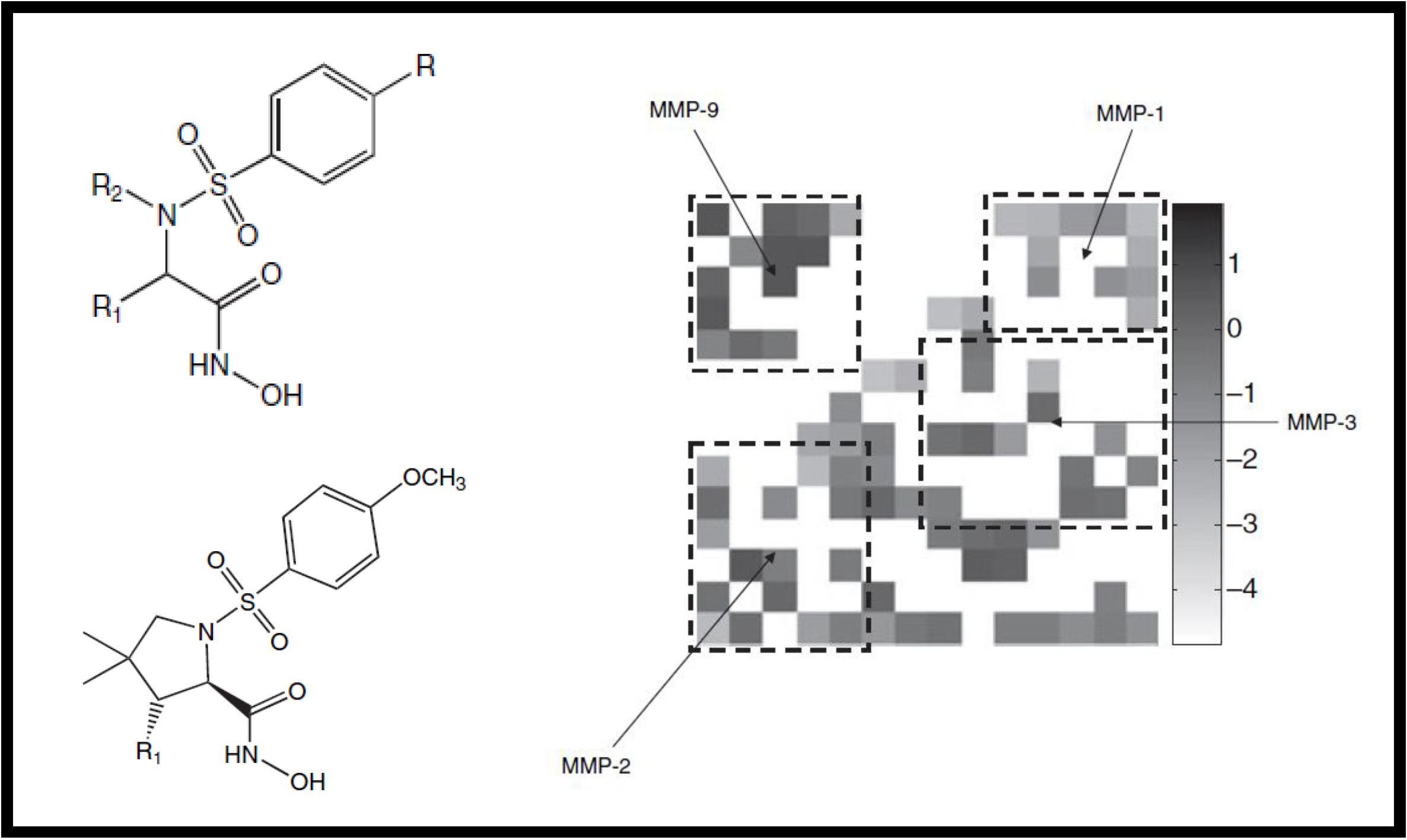
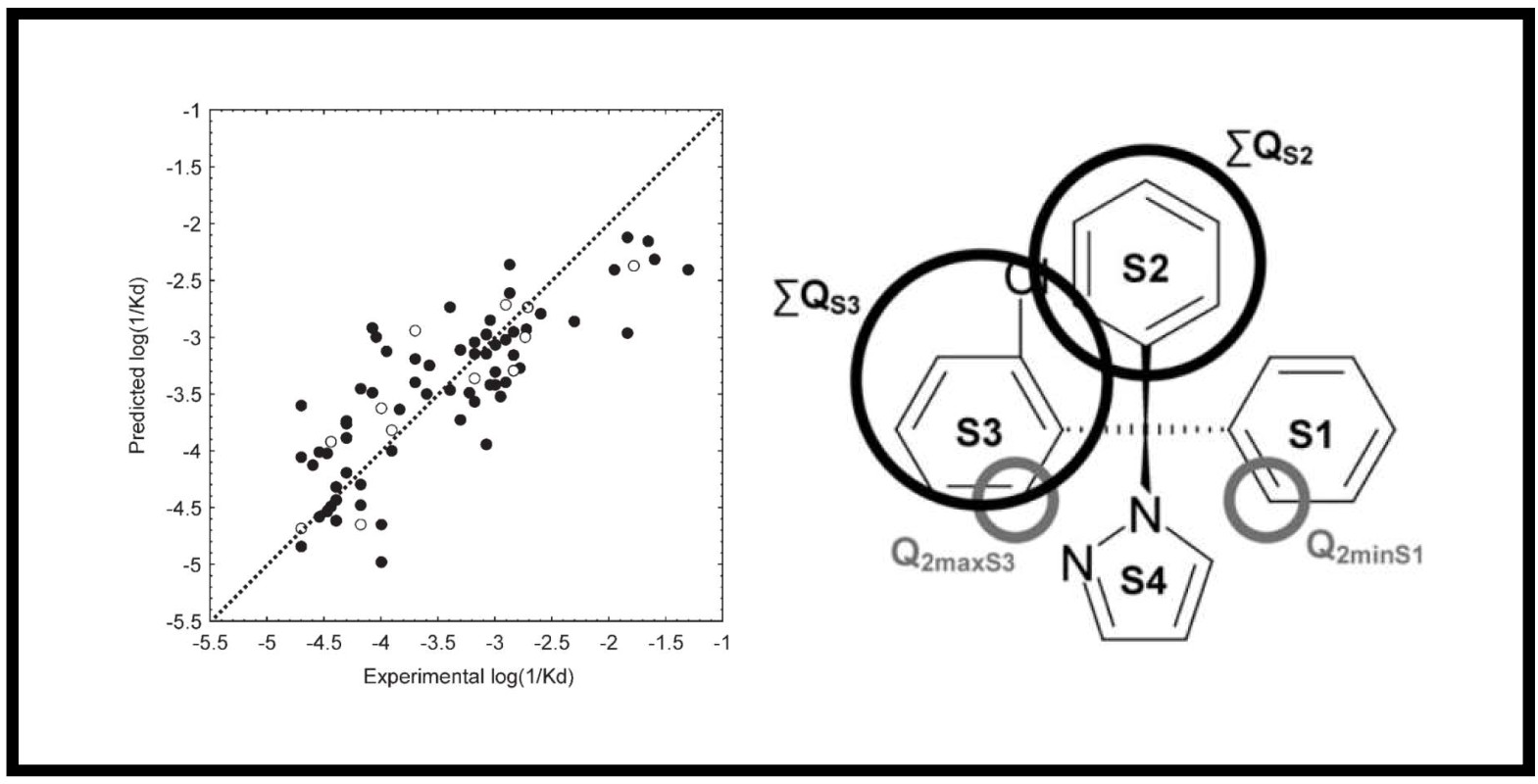
32.
M. Fernández*, J. Caballero, L. Fernández, J.I. Abreu, M. Garriga
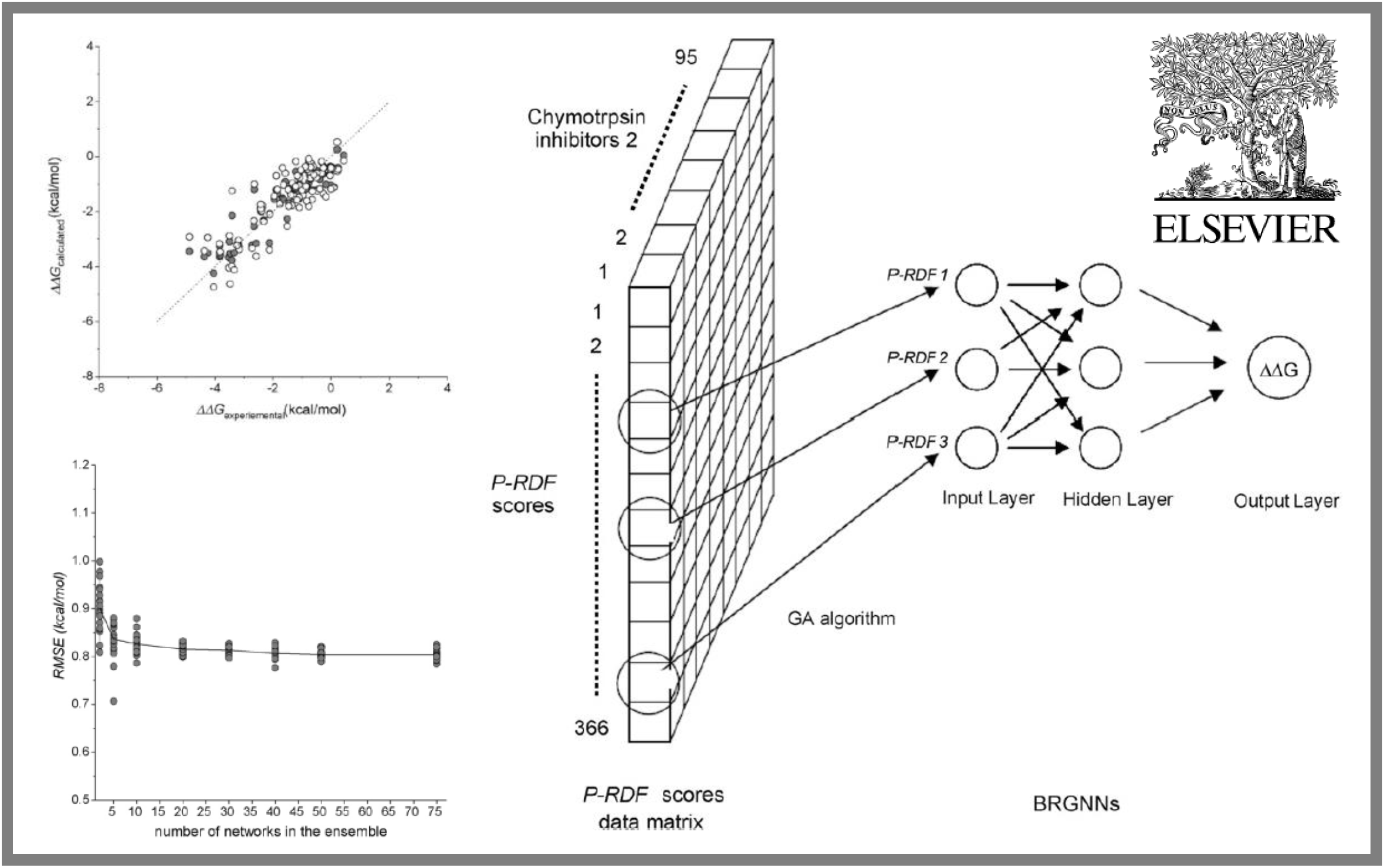

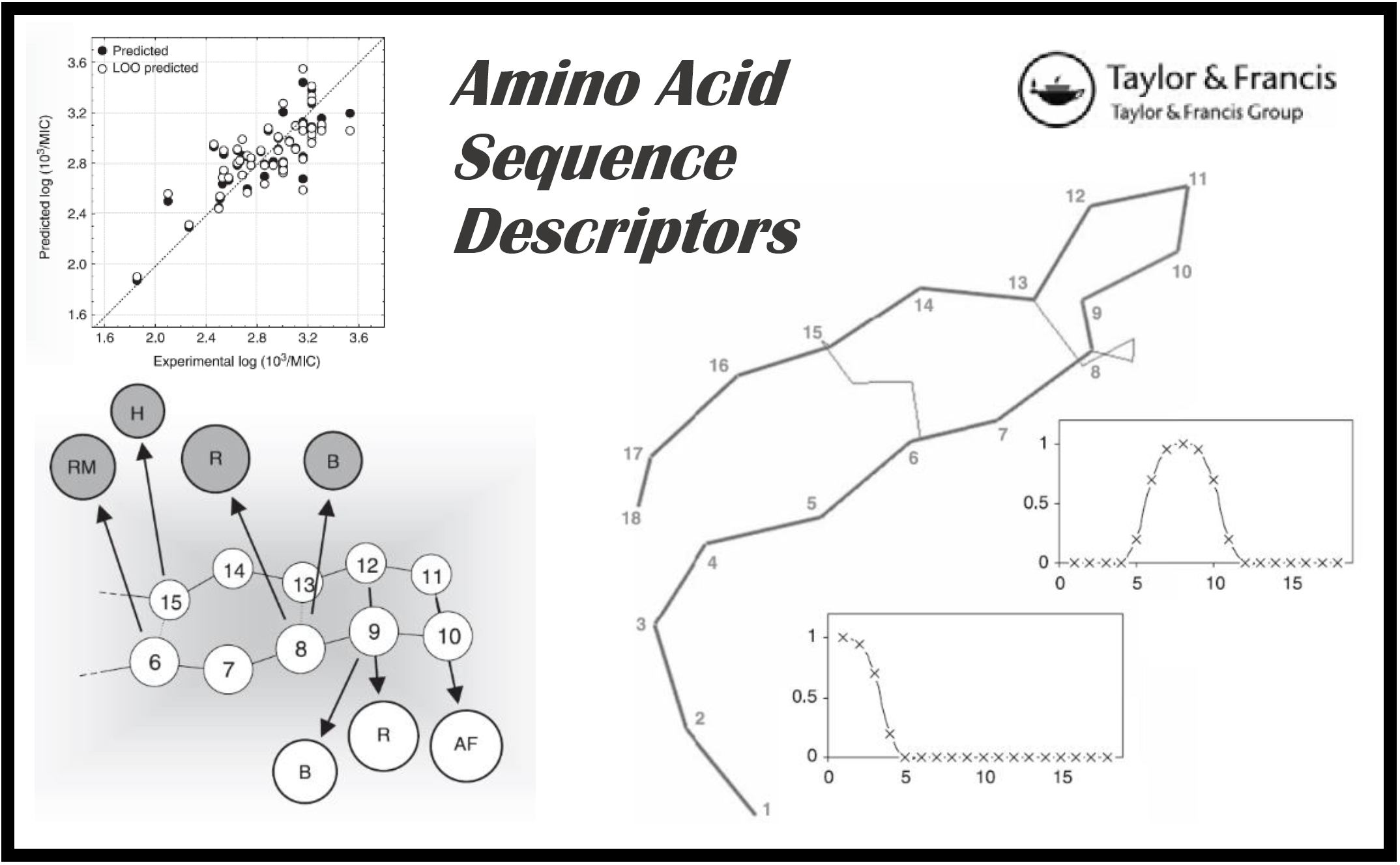
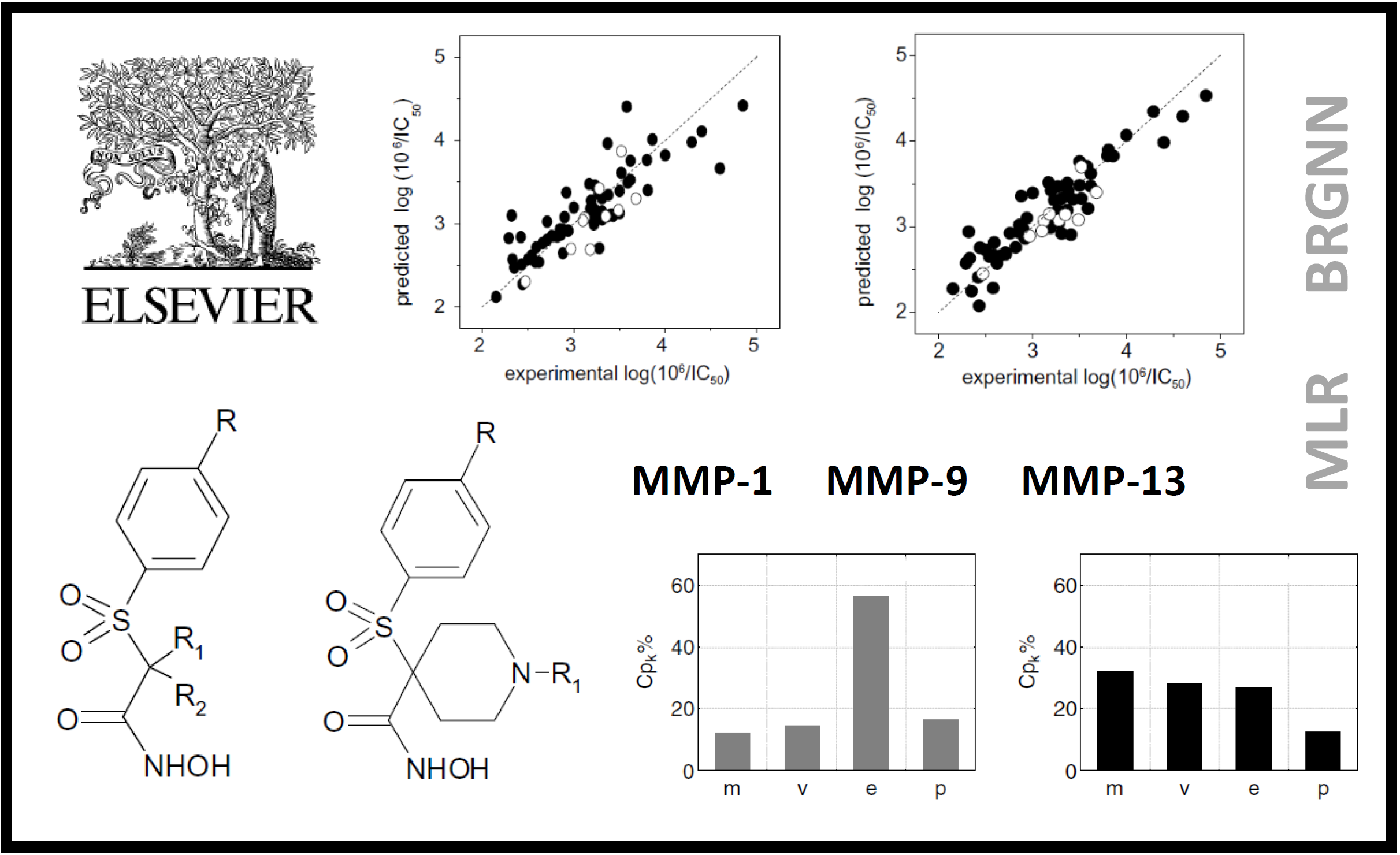
27.
J. Caballero, L. Fernández, M. Garriga, J. I. Abreu, S. Collina, M. Fernández*
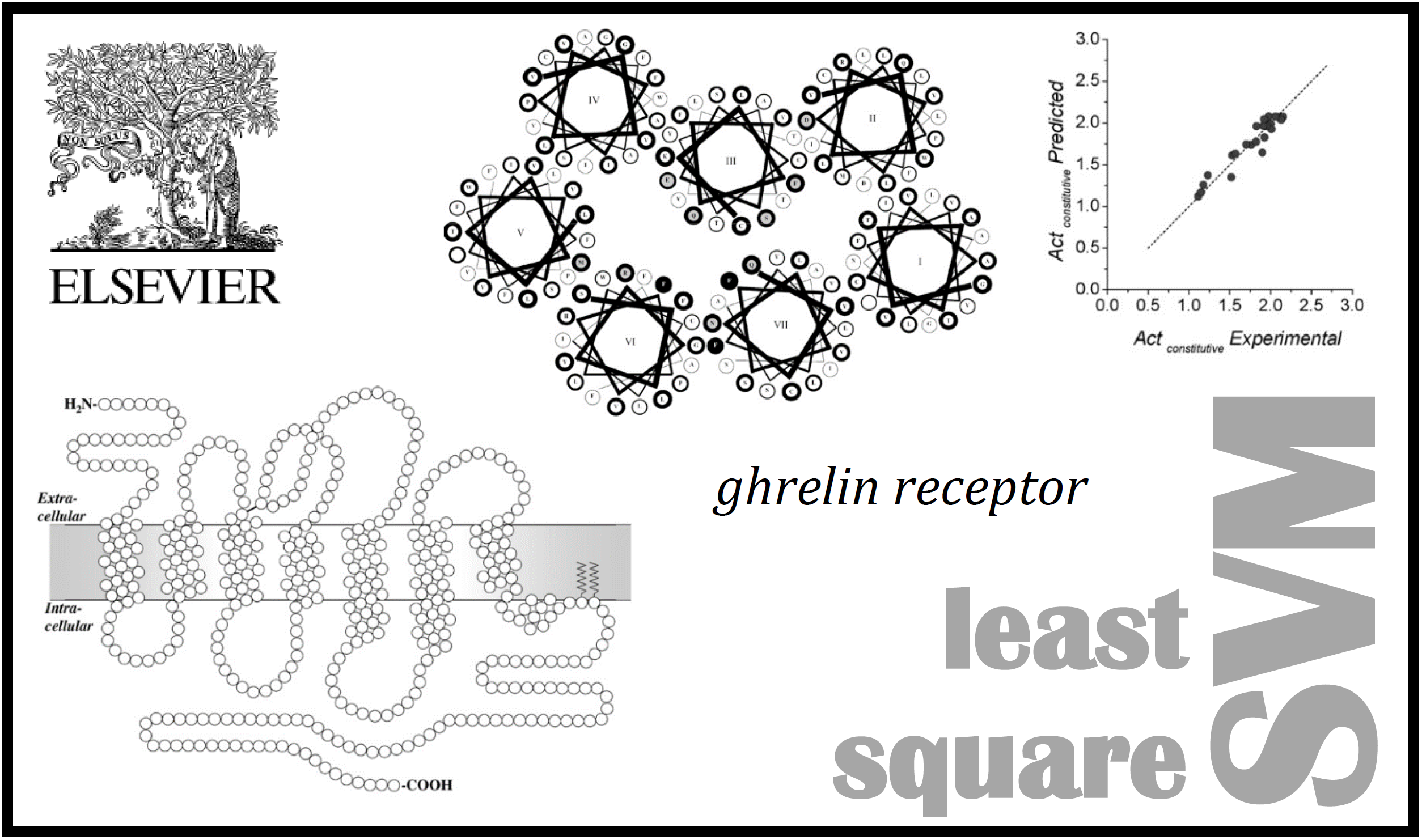
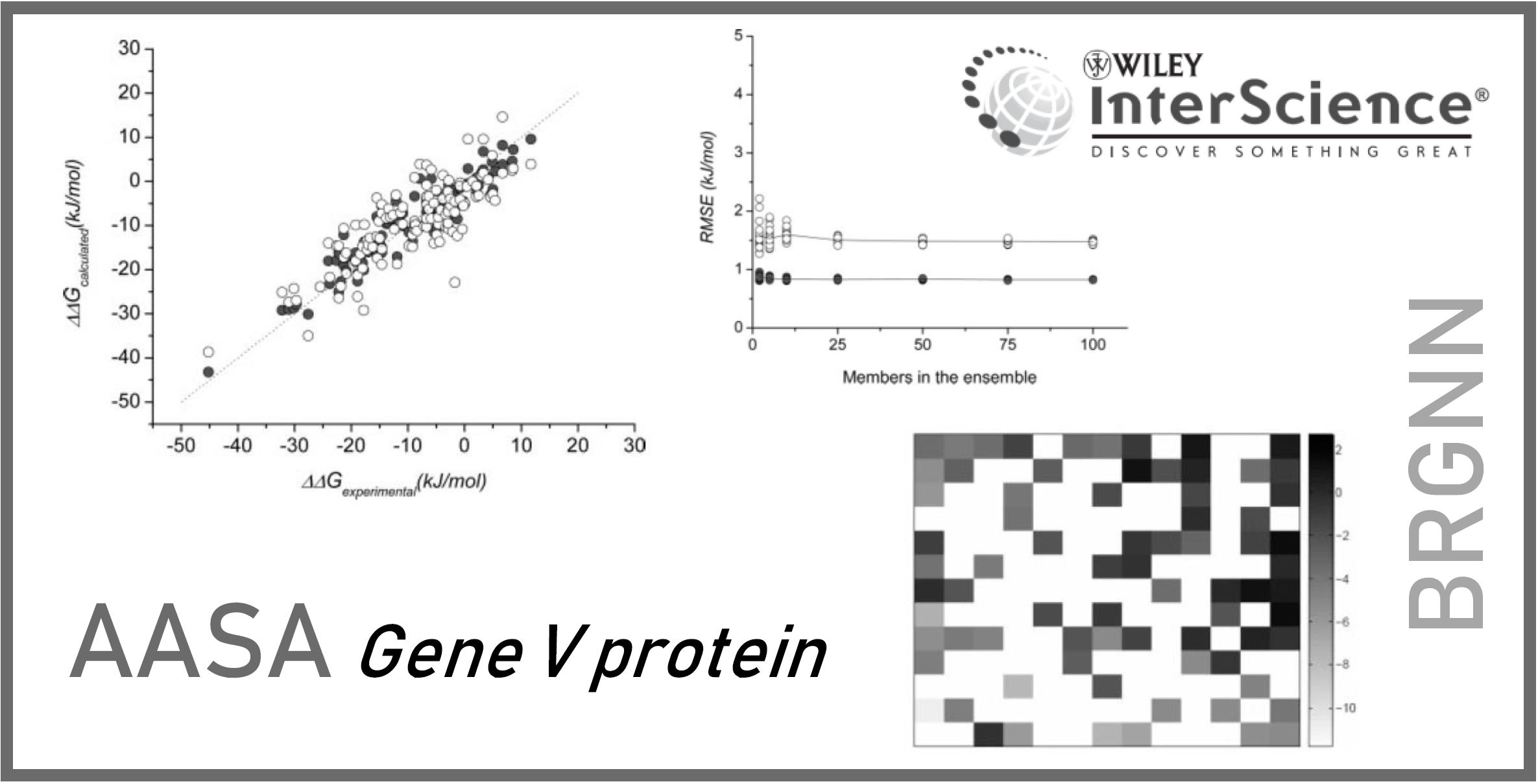
25.
P.R. Duchowicz*, M.G. Vitale, E.A. Castro, M. Fernández, J. Caballero
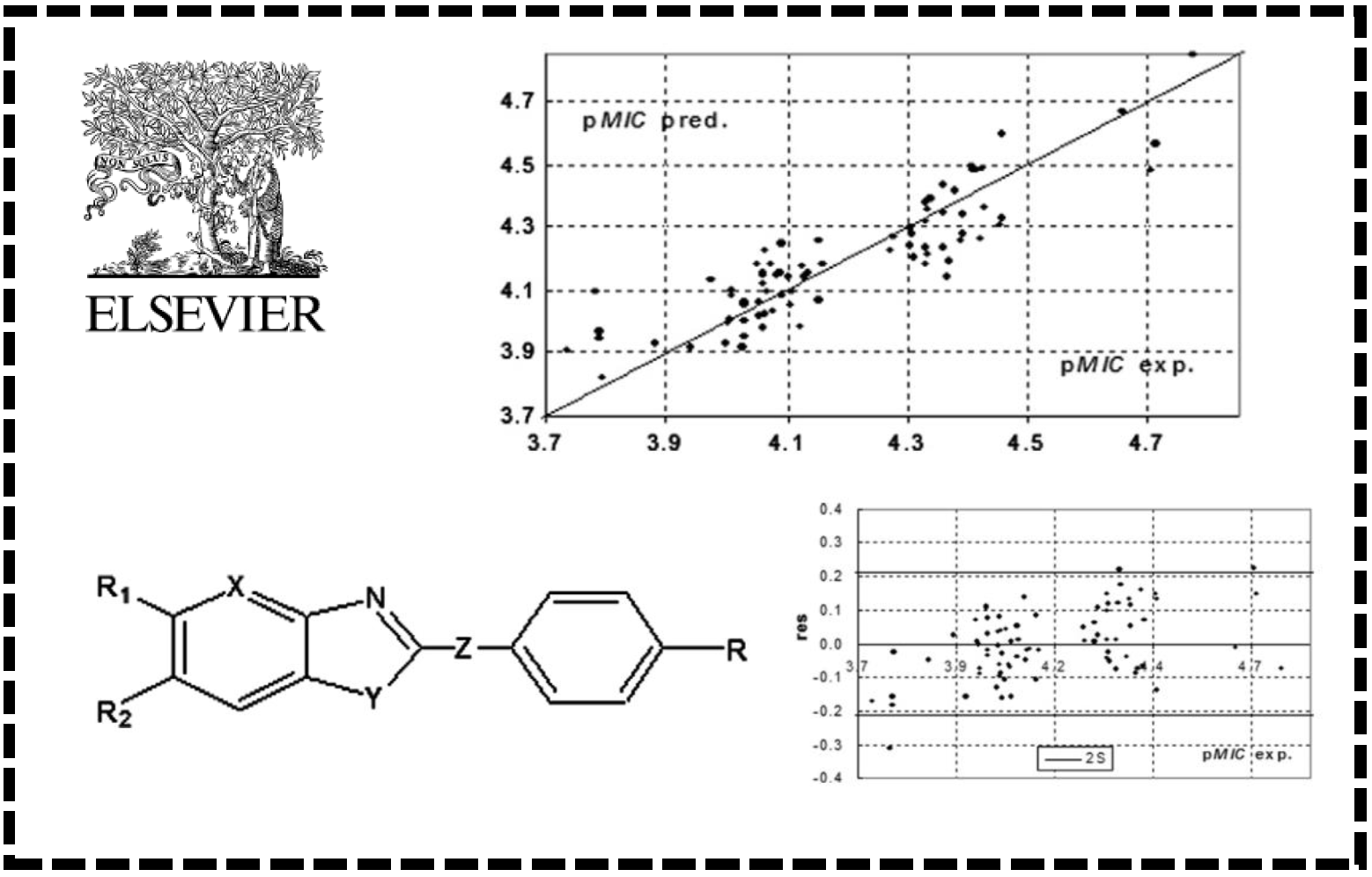
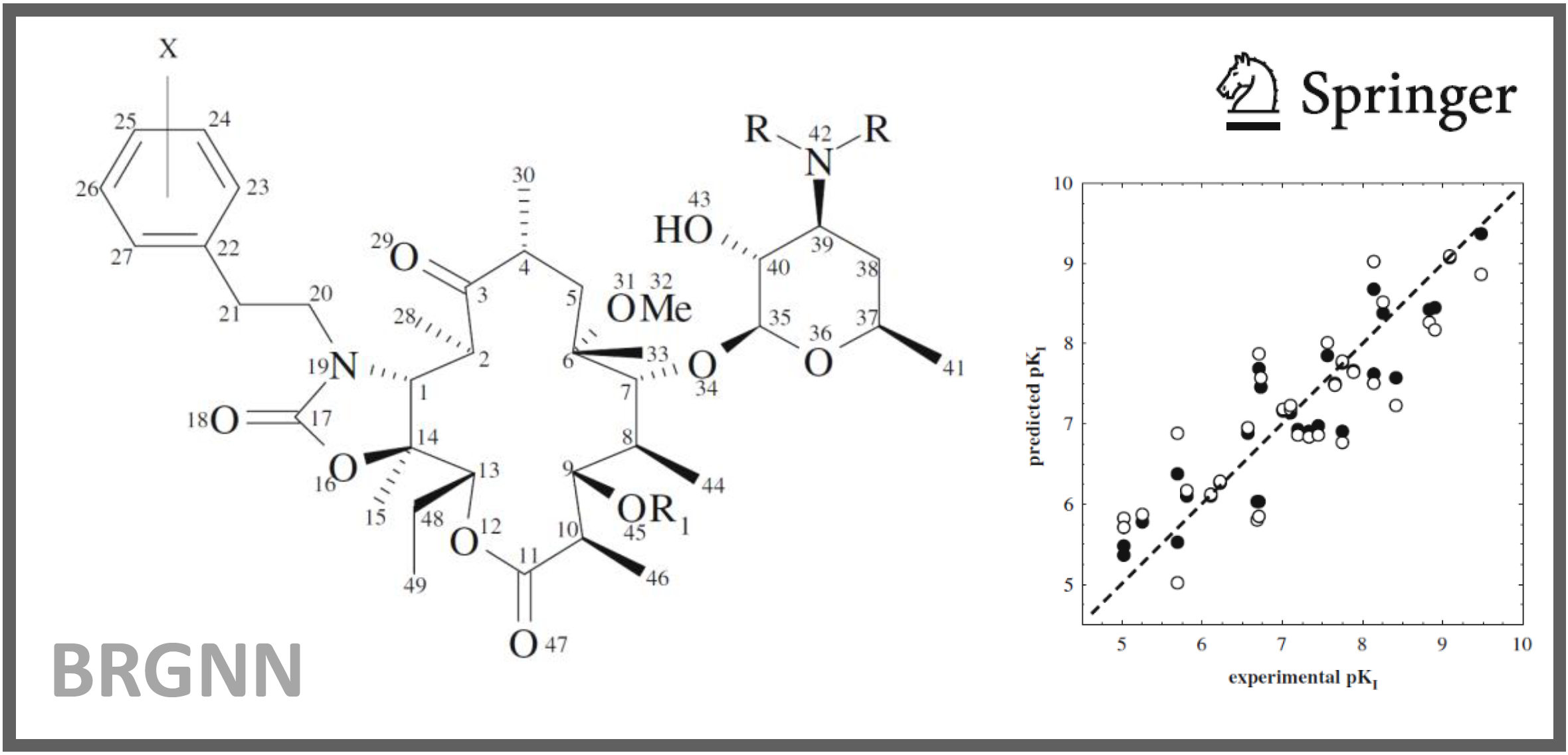
19.
P.R. Duchowicz*, M. Fernández, J. Caballero, E.A. Castro, F.M. Fernández
18.
M.P. González, J. Caballero, A.M. Helguera, M. Garriga, G. González, M. Fernández*
12.
M.P. González, J. Caballero, A. Tundidor-Camba, A.M. Helguera, M. Fernández*

10.
A. Valdivia, Y. Pérez, A. Domínguez, J. Caballero, Y. Hernández, R. Villalonga*
6.
M. Fernández, J. Caballero, A.M. Helguera, E.A. Castro, M.P. González*
5.
A. Domínguez, A. Valdivia, J. Caballero, R. Villalonga*, G. Martínez, E.H. Schacht
4.
R. Villalonga*, R. Cao*, A. Fragoso, A. Damiao, P. Ortiz, J. Caballero
3.
A. Valdivia, Y. Pérez, A. Domínguez, J. Caballero, L. Gómez, E.H. Schacht, R. Villalonga*
2.
M. Fernández, M.L. Villalonga, J. Caballero, A. Fragoso, R. Cao, R. Villalonga*